sMy research on peptides is focused on those that are able to form secondary structure elements in solution like 
In a first study, we have investigated the properties of a small
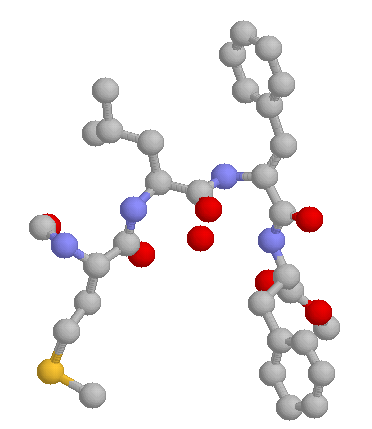
FIgure 1: the peptide For-Met-Leu-
The simplest case of the ß-sheet structure is the ß-hairpin. In the group of Prof. H.J.C. Berendsen at the University of Groningen, I have studied the 41-56 ß-hairpin from the protein G B1 domain [2] and subsequently, in collaboration with Giorgio Colombo (CNR milano, Italy) and Alan Mark synthetic ß-hairpins [3]. These studies have evidenced the role of the hydrophobic interactions residues in the stabilization of these peptides in aqueous solutions. In particular, the ß-hairpin containing a natural hydrophobic cluster from the protein G B1 and a D-Pro-Gly turn forming sequence have shown the role of diagonal interactions in driving the peptide to the correct fold structure and in keeping it in the ensemble of folded conformations. The combination of the stabilizing effects of the D-Pro-Gly turn sequence and of the hydrophobic nucleus formation thus favours the attainment of an ordered secondary structure compatible with the one determined experimentally. Moreover, our data underline the importance of the juxtapositions of the side chains of amino acids not directly facing each other in the three-dimensional structure. The combination of these interactions forces the peptide to sample a non-random portion of the conformational space, as can be seen in the rapid collapse to an ordered structure in the refolding simulation, and show that the unfolded state can be closely correlated to the folded ensemble of structures, at least in the case of small model peptides.
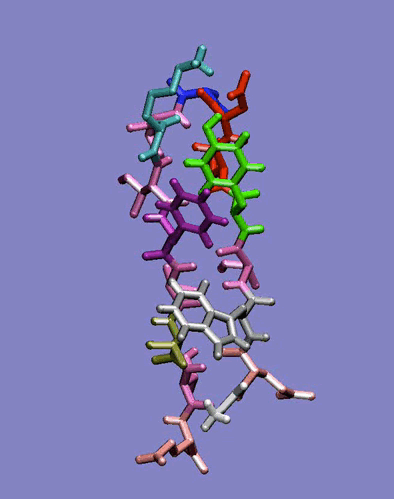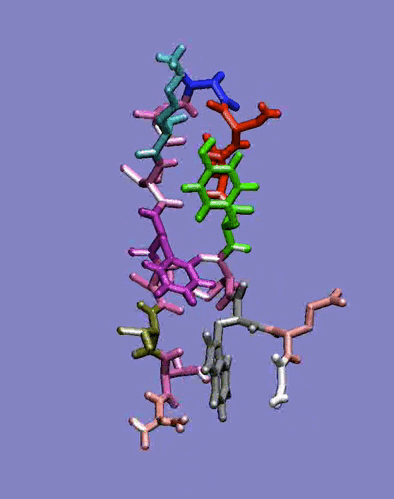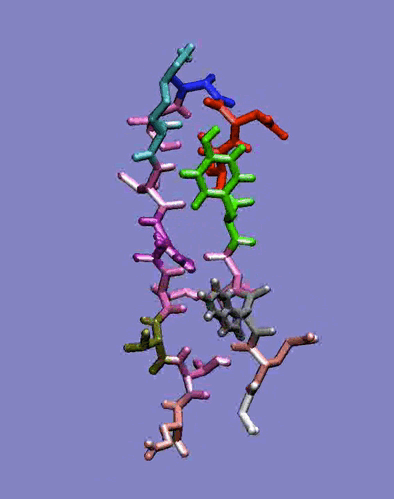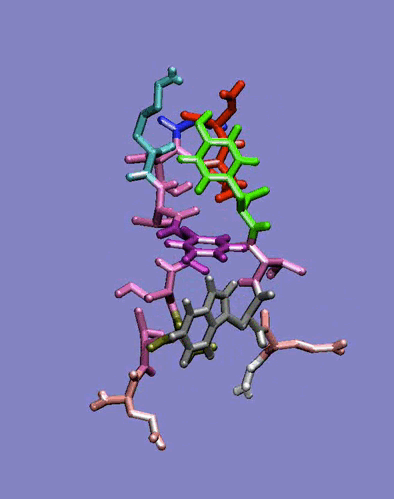
Figure 1: Dynamics in solution of the GB1 41-56 ß-hairpin from MD simulation (30 ns).
The other simpler ß-structure that was found to be from by peptide in solution is the three-stranded ß-sheet. In collaboration with Giorgio Colombo and Alan Mark, we have studied the dynamics of the three-stranded ß-sheet peptide Betanova at different temperatures by MD simulations in water [4]. Two 20 ns simulations at 280K indicate that the peptide remains very flexible under folding conditions sampling a range of conformations that together satisfy the NMR-derived experimental constraints. Two simulations at 300 K (above the experimental folding temperature) of 20 ns each show the partial formation of inactive like structure, which also satisfies most of the NOE constraints at 280 K. At higher temperature, the presence of compact states, in which a series of hydrophobic contacts remain present, are observed. This is consistent with experimental observations regarding the role of hydrophobic contacts in determining the peptide’s stability and in initiating the formation of turns and loops.
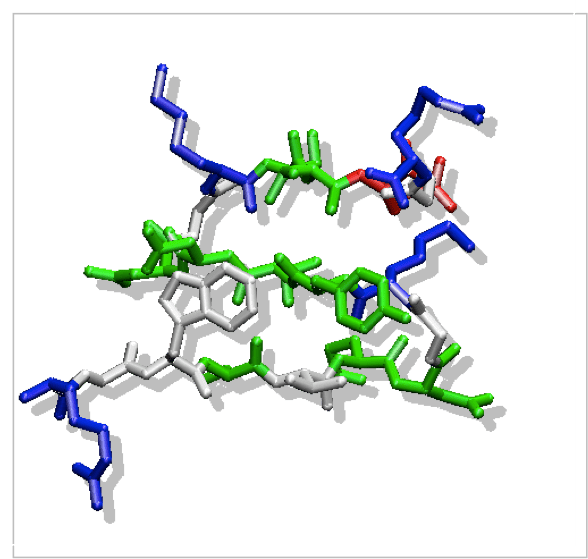
Figure 2: NMR structure of the Betanova peptide.
- A. Di Nola, E. Gavuzzo, F. Mazza, G. Pochetti, D. Roccatano. Internal β-turn hydration: crystallographic evidence and molecular dynamics simulation. J. Phys. Chem., 99, 9625-9631 (1995).
- Roccatano, A. Amadei, A. Di Nola, H. J. C. Berendsen. A molecular dynamics study of the 41-56 β-hairpin from B1 domain of Protein G. Protein Sci., 8, 2130-2143 (1999).
- Colombo, G. M. S. De Mori, D. Roccatano. Interplay between hydrophobic cluster and loop propensity in β-hairpin formation: a mechanistic study. Prot. Sci., 12, 538-550 (2003).
- Colombo, D. Roccatano, A. E. Mark. Folding and stability of the three-stranded β-sheet peptide betanova: Insights from molecular dynamics simulations. Proteins: Struct., Funct., Genet., 46, 380-392 (2002).

