In the previous article, we have learned how to set up the Hückel determinant for an aromatic molecule based on the topology of the pi-bonds. In this second part, we are going to learn how to calculate from the determinantal equation both the eigenvalues and the eigenvectors, corresponding to the orbital energy and orbital functions of the molecular system.
EIGENVALUES FROM THE HÜCKEL DETERMINANT
The calculation of the determinant gives a so-called characteristic equation. Namely, a polynomial equation whose roots (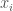


that is solved as
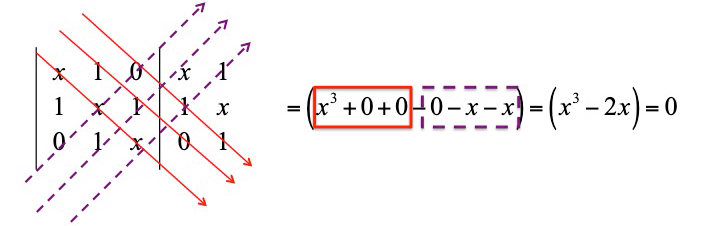
The polynomial 
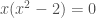


We can now use these value to calculate the energy of the Hückel orbitals using the relation
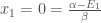




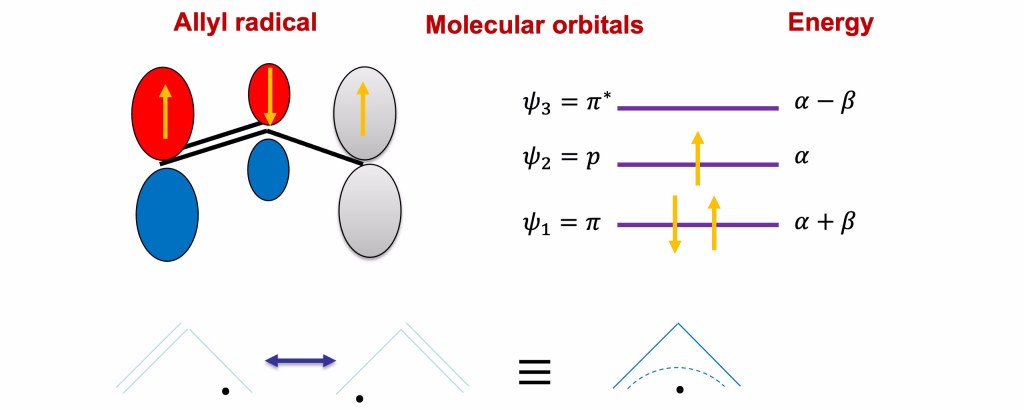

Calculation of Molecular Orbitals Coefficients
We are going to calculate the wavefunctions of the Hückel’s orbitals using the eigenvalues obtained from the characteristic equation. The coefficients of the molecular orbitals are calculated by solving the system of equations and the normalization condition given in the following slides.

For 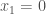



For 

Finally, the eigenfunctions and their graphical representation are reported in the following slide.

For linear alkenes, the eigenvalues and eigenvectors can be calculated using simple formulas reported in the following slide [1].

Here, the value for the allyl molecule is recalculated again using the formulas in the previous slide. A simple program, written in awk language is also listed in the appendix.

The numerical representation obtained by the formula is given as follows
Number of atoms : 3 Number of electrons : 2 Number of double occupied orbitals : 0 Number of single occupied orbitals : 0 Orbital Energies: E1=alpha -1.414beta E2=alpha -0.000beta E3=alpha +1.414beta Total Electronic Energy: Epi= 2.000alpha +2.828beta Wavefunctions: Psi(1)= +0.500 X(1) +0.707 X(2) +0.500 X(3) Psi(2)= +0.707 X(1) +0.000 X(2) -0.707 X(3) Psi(3)= +0.500 X(1) -0.707 X(2) +0.500 X(3)
The linear chain next to the allyl molecule is the 1,3-butadiene, composed of 4 carbon atoms and two conjugated double bonds—the Hückel topological matrix is shown in the next slide. The determinant gives a 4th-degree characteristic equation whose roots can easily be found by variable substitution, as shown in the slide.
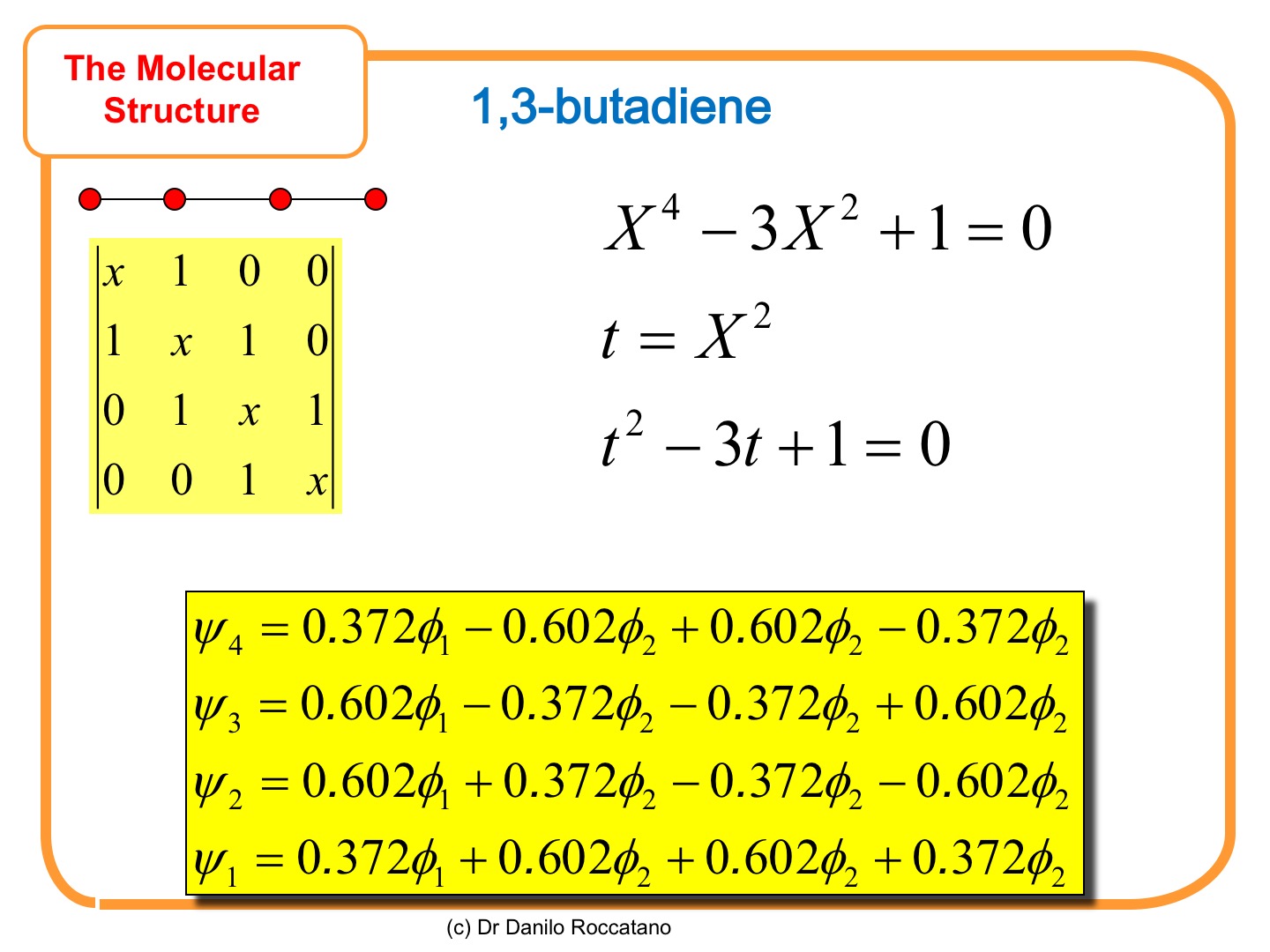
By running the program in the appendix, we can find the value of the energy and of the coefficients of the wavefunction
Number of atoms : 4 Number of electrons : 4 Number of double occupied orbitals : 0 Number of single occupied orbitals : 0 Orbital Energies: E1=alpha -1.618beta E2=alpha -0.618beta E3=alpha +0.618beta E4=alpha +1.618beta Total Electronic Energy: Epi= 4.000alpha +4.472beta Wavefunctions: Psi(1)= +0.372 X(1) +0.602 X(2) +0.602 X(3) +0.372 X(4) Psi(2)= +0.602 X(1) +0.372 X(2) -0.372 X(3) -0.602 X(4) Psi(3)= +0.602 X(1) -0.372 X(2) -0.372 X(3) +0.602 X(4) Psi(4)= +0.372 X(1) -0.602 X(2) +0.602 X(3) -0.372 X(4)
A rotational barrier separates the s-cis- and s-trans conformers of the 1,3-Butadiene. We can try to make an estimation of the transition barrier using the HMO (see Ref. [2]). We can assume that the two double bonds are not conjugated during the rotation as for the allyl molecules. Therefore, the energy of the transition state is given by the sum of the two isolated double bonds’ energy. Being a total of four electrons in the 


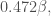



Even though the HMO method is simple and approximated, it provided crucial results to understand the organic reaction mechanism. In the next article, we will calculate the HMO for cyclic conjugated molecules and discuss other results of this method.
If you have found an interesting and useful article, do not forget to press “Like it” and subscribe for updates on new ones!
REFERENCES
- J. P. Lowe. Quantum Chemistry. 1993, Academic Press.
- F.A. Carroll. Structure and Mechanism,1998, BROOKS/COLE Publishing Company.
APPENDIX
Program in awk language to calculate the energy and coefficients of the Hückle orbitals. You may change the variable for the number of atoms (n) and the number of electrons (ne) to your convenience before running the program using the command:
gawk -f HMOLinearMol.awk
#======================================================================
#
# NAME: HMOLinearMol.awk
#
#======================================================================
# DESCRIPTION: Calculate the energy level and coefficients of the
# wavefunctions
#======================================================================
# Copyright (C): 2021 Danilo Roccatano
#======================================================================
#
BEGIN {
n=4 # number of atoms in the chain
ne = 5 # number of electrons in the chain
pi=4*atan2(1,1)
#
printf "Number of atoms : %d \n",n
printf "Number of electrons : %d \n",ne
printf "Number of double occupied orbitals : %d \n",npe
printf "Number of single occupied orbitals : %d \n",nue
# Calculate the eigenvalues
tEa=0
tEb=0
printf "Orbital Energies:\n"
for (k=1;k<=n;k++) {
x=-2*cos(k*pi/(n+1))
printf " E%d=alpha %+8.3fbeta\n",k,x
Eb[k]=x
}
#
# Calculate the total energy of the electronic system
#
npe=int(ne/2)
nue=ne-2*npe
for (i=1;i<=npe+nue;i++) {
fa=1
if (i<=npe) fa=2
tEa+=fa
tEb+=fa*Eb[n-(i-1)]
}
printf "Total Electronic Energy:\n"
printf " Epi=%8.3falpha %+8.3fbeta\n",tEa,tEb
#
# Calculate the wavefunction coefficients
#
printf "Wavefunctions:\n"
for (i=1;i<=n;i++) {
printf " Psi(%d)= ",i
for (k=1;k<=n;k++) {
c[k]=sqrt(2/(n+1))*sin(k*i*pi/(n+1))
printf "%+8.3f X(%d)",c[k],k
}
printf"\n"
}
}


Pingback: Physical Chemistry: The Simple Hückel Method (Part V) |
Pingback: Physical Chemistry: The Simple Hückel Method (Part VI): PREVIEW |