The Hückel molecular orbital method is a quantum mechanics approach for calculating the energies of molecular orbitals of π electrons in conjugated hydrocarbon systems, such as ethylene, benzene, and butadiene. In this series of articles, I have summarized the main aspects of the theory with practical examples of applications and programming of the method.
The method was devised by Erich Hückel in 1930 and subsequently expanded and improved by other scholars. The technique has helped obtain important theoretical results in studying of the properties of organic molecules and the mechanisms of their chemical reactions in the period preceding the development of electronic computers. For example, he provided the theoretical basis of Hückel’s rule for the aromaticity of (4n + 2) π cyclic planar systems of electrons.
Hückel assumed that the electrons in 

Hückel assumed that the π electrons can be treated separately by those involved in the
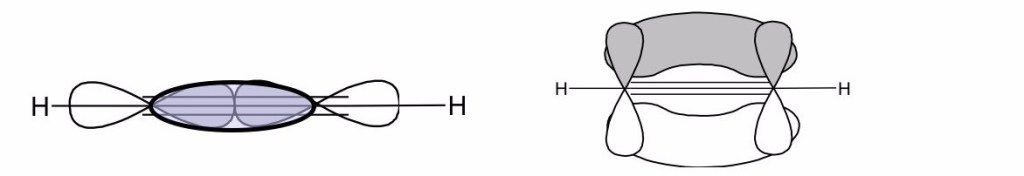
With such an assumption, we can then write the total energy of the molecular system as 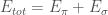
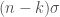


We can also assume that the molecular wave function of the whole system can be approximated as a product of 1-electron wave function orbitals

The energy of a sysmt can be evaulated using tha Hamiltonian quantum operator that is defined as

with
: the kinetic energy of the nucleus.
: the kinetic energy of the electrons.
: the proton-electron attraction potential energy.
: the proton-proton repulsion potential energy.
: the electron-electron repulsion energy.
In the case of a molecule composed by M atoms, the total hamiltonian can be written as
For a molecular sytem with π electrons, we can further distinguish these electron from the

For not interaction electrons, we can also assume that the total energy of the system can be calculate by a total Hamiltonian (see my blog on the classical mechanics) operator can be expressed as


and the eigenstates of each electron can be calculate by the Schrödingen’s equations

As further assumption the wavefunction of the 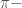
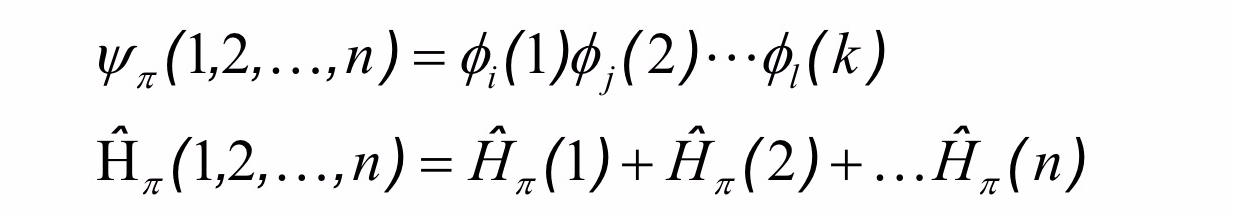
Using this approximations the total electronic energy from the
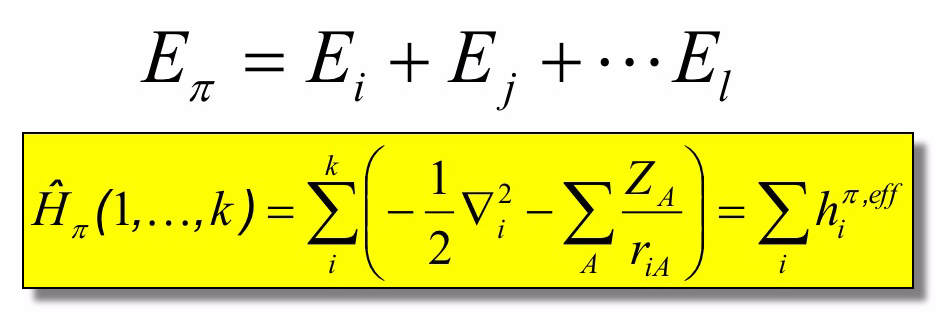
We are now going to consider a simple molecular system (the allyl molecule) to show how to assign the atomic orbitals to construct the Hückel determinant. An allyl molecule has the structural formula 


We consider only the three orbitals 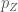
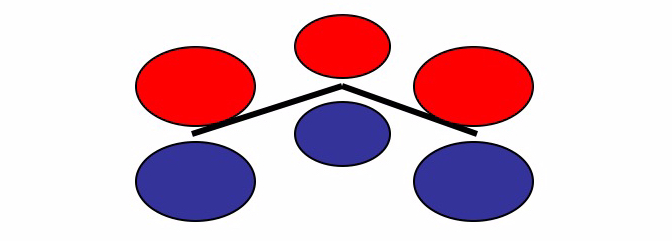
The total molecular orbital can be considered as a linear combination of the atomic orbitals.
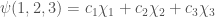
In order to find the energy of the molecular system, we are going to use the result of the so called variational theorem.
The variation theorem states that given a system with a Hamiltonian operator 
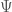

where 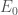

The variational principle allows us to calculate an upper bound for the ground state energy by finding the trial wavefunction 

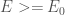
Linear variation method
We are going to use the so-called linear variation method (or Rayleigh-Ritz method) in which the linear variation function is a linear combination of 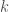
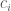
By inserting the function
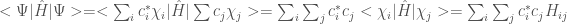
where 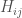
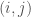

where 
Therefore we can now write

or in the rearranged form as

Now, we need to find the value of the coefficients 
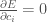
for all the functions i.
By differentiating both the sides of (1) and using the properties of

for all the k.
We therefore have a set of k simultaneous homogeneous equation (secular equations) in k unknowns. In the case of the allyl system the system is the follwing
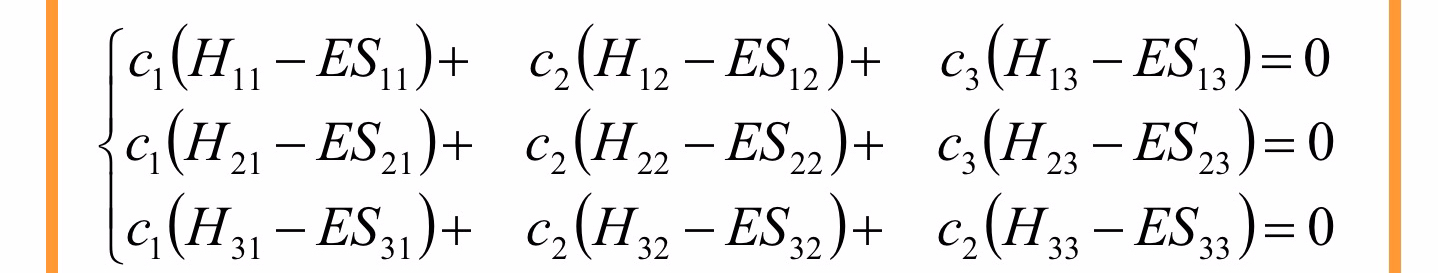
For a non-trivial solution (i.e. 
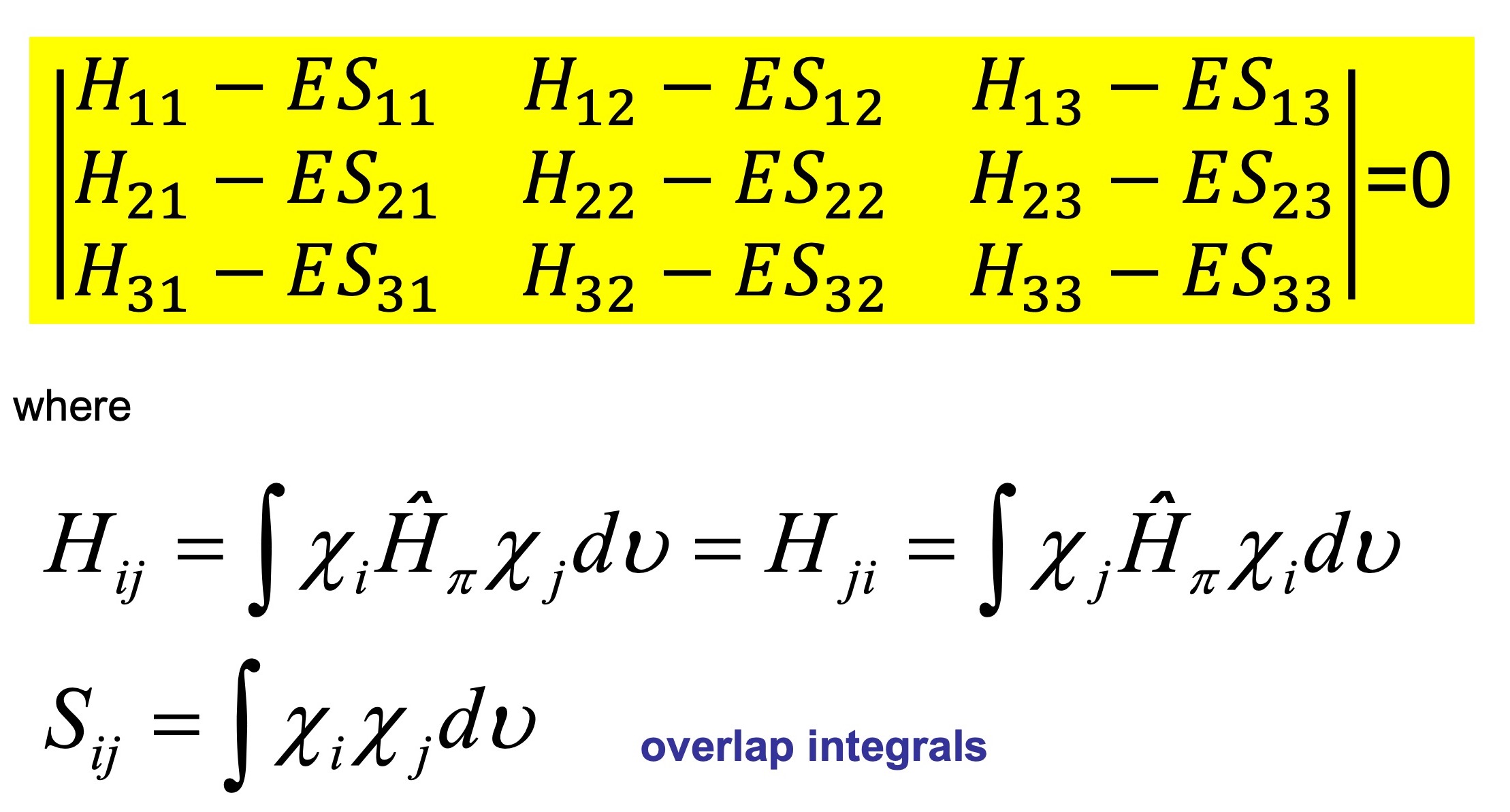

Therefore we can set up the values of

and for the overlap integral
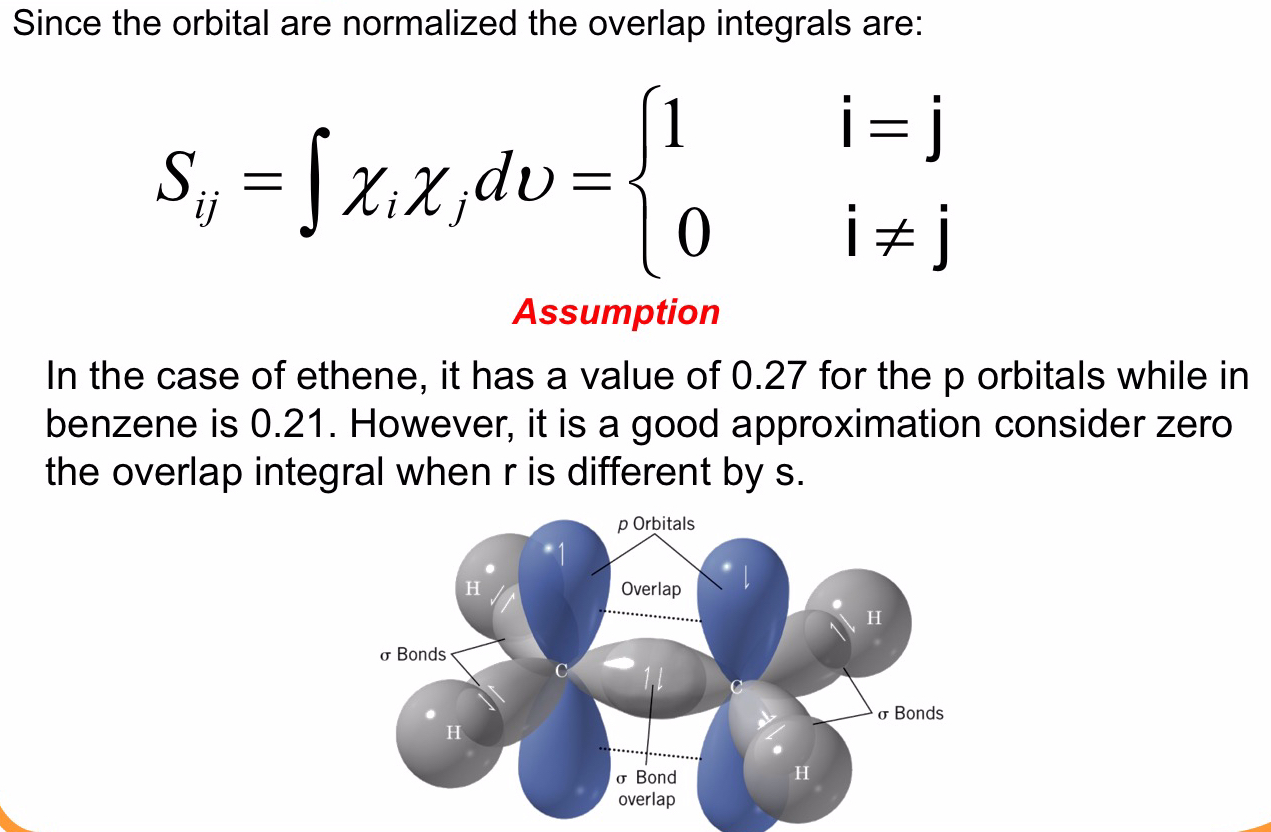
With these semplification the Hückel determinant for the allyl molecule become


Here some more example of Hückel determinat for simple unsaturated molecules
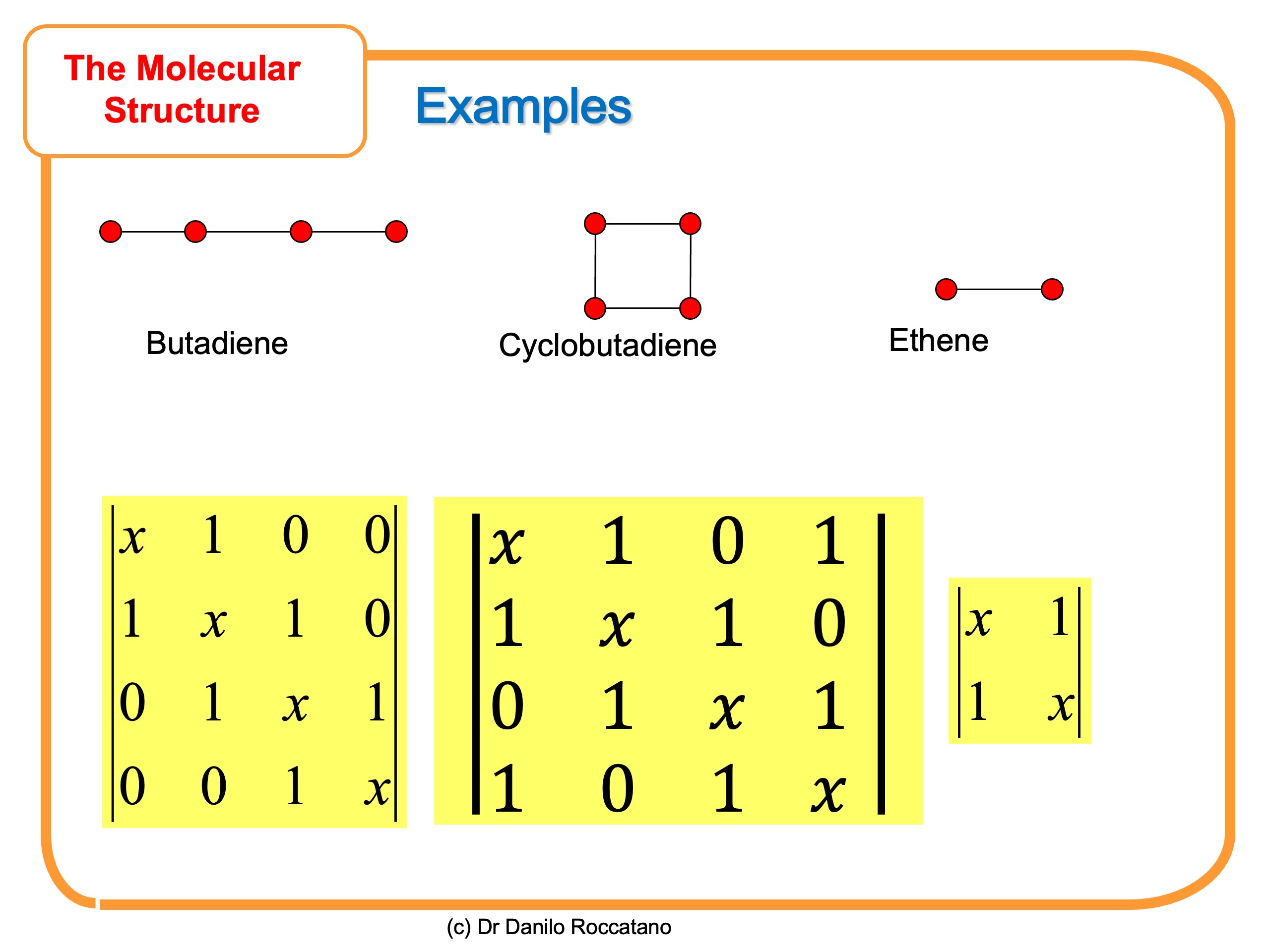
In the second part of this article, we are going to learn how to solve this determinantal equation to find the energy of the molecular orbitals.
If you have found useful this article, please tell your friends and do not forget to Like it and subscribe for updating!
REFERENCES
J.P. Lowe Quantum Chemistry. 1993, Academic Press.


Pingback: Physical Chemistry: The Simple Hückel Method (Part V) |
Pingback: Physical Chemistry: The Simple Hückel Method (Part VI): PREVIEW |
Hello,
first thing thank you for the interesting articles on your web site.
Reading the Hückel Method (part I) I’ve spotted some mistakes.
In the sentence: ” In fact, a \pi orbital is antisymmetric by reflection on the molecule plane, while a $ latex sigma $ is symmetrical.” reported at the end of the third paragraph, the HTML page doesn’t show the sigma character but its latex code instead.
in slide11-e1605347034130.jpeg the error is that:
the second term in the first row of Hückel determinant for a three p(z) orbitals system is the same as the first term of the first line. It should be H12 – ES 12.
and
in slide17.jpeg there is an error in the Hückel determinat for cyclobutadiene
The correct determinant should be:
| x 1 0 1 |
| 1 x 1 0 |
| 0 1 x 1 |
| 1 0 1 x |
I hope this could help improve the quality of your web page.
Cheers,
alessandro
LikeLike
Hi Alessandro,
Thank you very much for reporting the mistakes. I am also happy to read that you find my articles interesting, and constructive comments are always welcome.
Cordiali saluti
Danilo
LikeLike