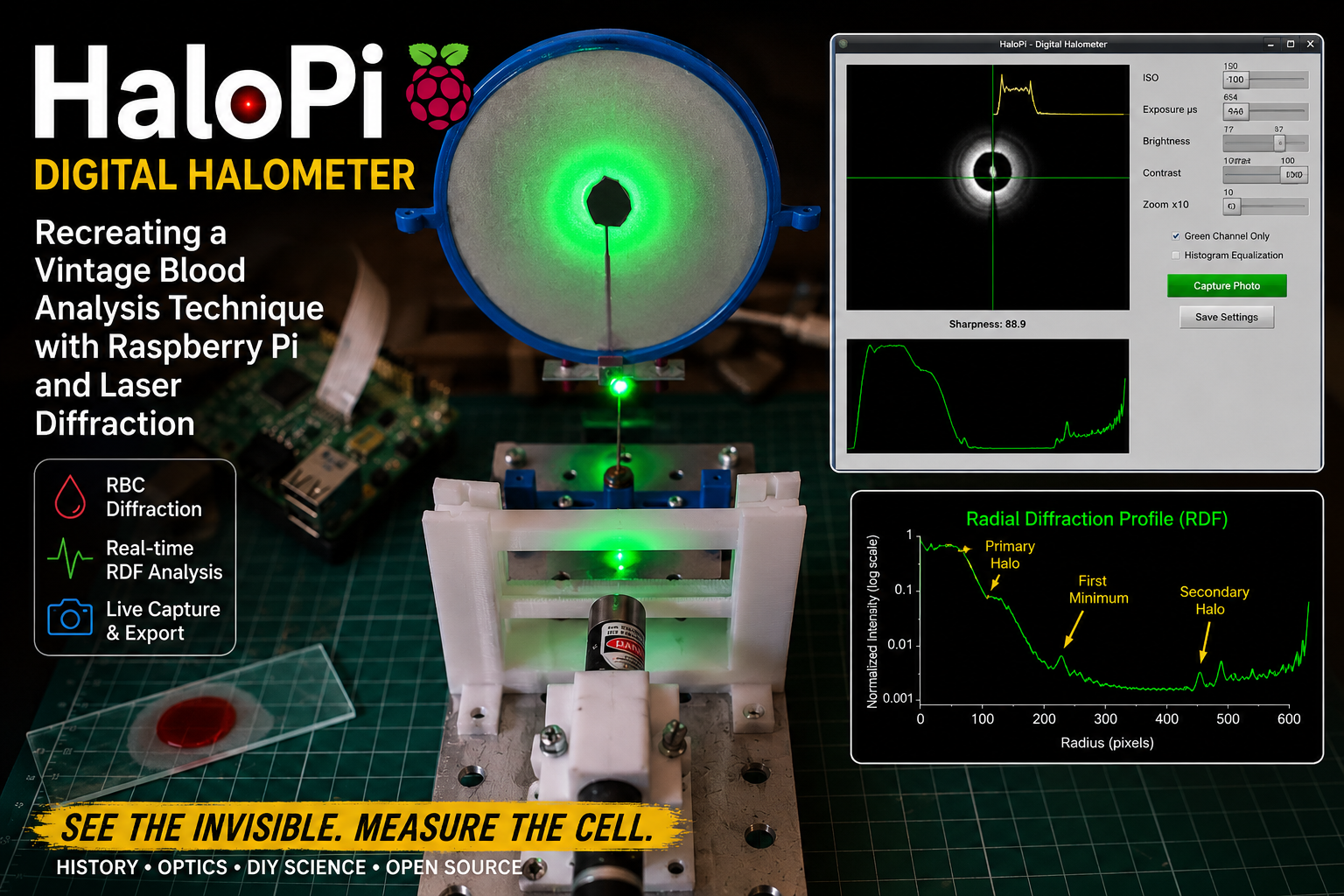
One of the aspects of experimental science that I find most fascinating is the rediscovery of historical scientific instruments and techniques that have gradually disappeared from modern laboratories. In my latest Instructable project, I explored the reconstruction of a remarkable optical device once used in hematology during the early twentieth century: the halometer.
Before the advent of automated blood analyzers and digital microscopy, researchers investigated ingenious indirect methods for estimating the average size of red blood cells. One of these methods relied on diffraction phenomena produced when coherent light passed through thin blood smears. The resulting circular halos could be related to the average diameter of erythrocytes, providing a rapid—although approximate—diagnostic technique for conditions such as pernicious anemia.