In the previous article, we have learned how to set up the Hückel determinant for conjugated linear molecules based on the topology of the 
The cyclopropenyl system: the cyclic equivalent of the allyl molecule
Cyclopropenyl is a well-studied system studied. The cation is generated by mixing 3-chlorocyclopropene with strong Lewis acids such as antimony pentachloride, aluminum trichloride, or silver fluoroborate. The salt is stable and can be studied using NMR [1].
The Hückel determinant for the cyclopropenyl cation is shown in the following picture.

The characteristic equation is the cubic polynomial

with roots

The cation is a two electrons system, therefore we can represent the orbital energy in this way

The coefficients of the orbital function are found by solving the system

Therefore, we obtain the coefficients for the wavefunction of the orbital with the lowest energy by setting the eigenvalue 

In the following slide, the wavefunction 

We can proceed finding a second set of coefficient by setting 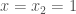
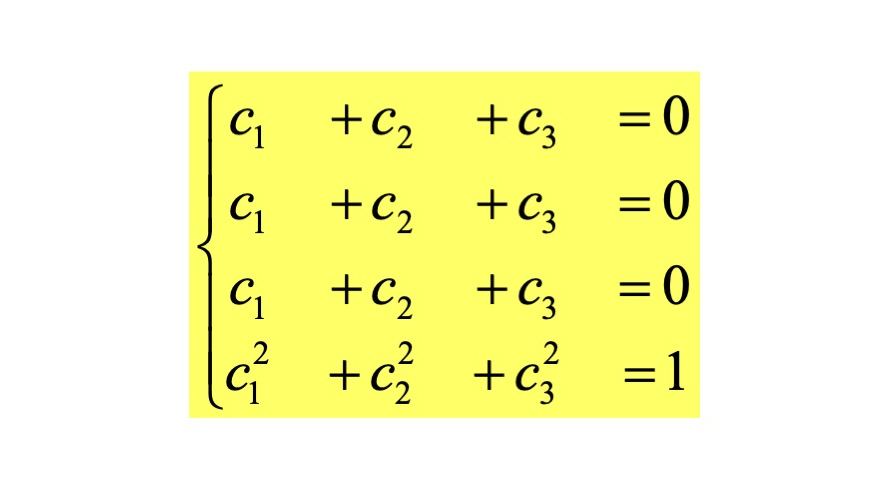
The equation are linear dependents and this give infinite possible solution. For convenience we set 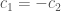


For the third solution, we need to find a new orthonormal wave function using a mathematical procedure know as Gram-Schmidt orthogonalization. This is accompished by imposing the condition of normalizzation starting from one set of coefficients


We define a function 
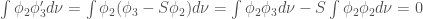
Therefore,

By renomarlization

The wavefunctions, 
It is important to note that the SHMO’s orbital level refers to symmetric molecules with equilateral structures. Indeed, this structure is unstable, and in reality, the structure is distorted to an isosceles triangle separating the energy level of the two degenerated states. That is an example of the Jahn-Teller theorem, which states that a system with an odd number of electrons in degenerated molecular orbitals will change its geometry to remove the degeneracy [2].

Also for cyclic polyene, there are the simple formulas reported in the following slides that can be used to calculate the energy and the coefficients of the HMO.

The output of the program for the cyclopropenyl system is the following
Number of atoms : 3 Number of electrons : 2 Number of double occupied orbitals : 0 Number of single occupied orbitals : 0 Orbital Energies: E1=alpha +2.000beta E2=alpha -1.000beta E3=alpha -1.000beta Total Electronic Energy: Epi= 2.000alpha +2.000beta Wavefunctions: Psi(1)= ( +0.577)X(1)( +0.577)X(2)( +0.577)X(3) Psi(2)= ( +0.000)X(1)( -0.707)X(2)( +0.707)X(3) Psi(3)= ( +0.816)X(1)( -0.408)X(2)( -0.408)X(3)
THE MOLECULE OF BENZENE
The benzene molecule is a 6 carbon ring. There are 6 electrons delocalized in 6 atomic pz orbitals that contribute to form the Hückel molecular orbitals, as shown in the next figure

The Hückel 6×6 determinant is shown in the left side of the next slide

The solution of the determinant produce 6 eigenvalues 

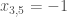

The total energy is equal to 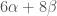



The coefficients of the MOs calculated using the program reported in the appendix are the following:
Number of atoms : 6 Number of electrons : 6 Number of double occupied orbitals : 0 Number of single occupied orbitals : 0 Orbital Energies: E1=alpha +2.000beta E2=alpha +1.000beta E3=alpha +1.000beta E4=alpha -1.000beta E5=alpha -1.000beta E6=alpha -2.000beta Total Electronic Energy: Epi= 6.000alpha +8.000beta Wavefunctions: Psi(1)= ( +0.408)X(1)( +0.408)X(2)( +0.408)X(3)( +0.408)X(4)( +0.408)X(5)( +0.408)X(6) Psi(2)= ( +0.000)X(1)( -0.500)X(2)( +0.500)X(3)( -0.000)X(4)( -0.500)X(5)( +0.500)X(6) Psi(3)= ( +0.577)X(1)( -0.289)X(2)( -0.289)X(3)( +0.577)X(4)( -0.289)X(5)( -0.289)X(6) Psi(4)= ( +0.000)X(1)( -0.500)X(2)( +0.500)X(3)( -0.000)X(4)( -0.500)X(5)( +0.500)X(6) Psi(5)= ( +0.577)X(1)( -0.289)X(2)( -0.289)X(3)( +0.577)X(4)( -0.289)X(5)( -0.289)X(6) Psi(6)= ( +0.408)X(1)( -0.408)X(2)( +0.408)X(3)( -0.408)X(4)( +0.408)X(5)( -0.408)X(6)
The following slide shows a pictorial representation of the signs of the p-orbital functions in each MO.

In the next article, we are going to use the Hückel MOs to calculate important molecular properties.
If you have found an interesting and useful article, do not forget to press “Like it” and subscribe for updates on new ones!
REFERENCES
- Breslow, R. and Groves, J.T., 1970. Cyclopropenyl cation. Synthesis and characterization. Journal of the American Chemical Society, 92(4), pp.984-987.
- J. P. Lowe. Quantum Chemistry. 1993, Academic Press.
- F.A. Carroll. Structure and Mechanism,1998, BROOKS/COLE Publishing Company.
APPENDIX
Program in awk language to calculate the energy and coefficients of the Hückel molecular orbitals. You may change the variable for the number of atoms (n) and the number of electrons (ne) to your convenience before running the program using the command:
gawk -f HMOCyclicMol.awk
#======================================================================
#
# NAME: HMOCyclicMol.awk
#
#======================================================================
# DESCRIPTION: Calculate the energy level and coefficients of the
# Huckel orbitals for cyclic polyenes
#======================================================================
# Copyright (C): 2021 Danilo Roccatano
#======================================================================
#
function bubble(n,x, ix) {
#
# Improved bubble sort method
#
for (i=1;i<=n-1;i++) {
b=x[i]
ib=ix[i]
k=i
for (j=i+1;j<=n;j++) {
if (b>=x[j]) {
b=x[j]
ib=ix[j]
k=j
}
}
x[k]=x[i]
x[i]=b
ix[k]=ix[i]
ix[i]=ib
}
}
function inprod(n,vec,vec1) {
inp=0.0
for (i=1;i<=n;i++ ) {
inp += vec[i]*vec1[i]
}
return inp
}
function abs(c) {return c<0?-c:c}
BEGIN {
n=3 # number of atoms in the ring
ne = 2 # number of electrons in the ring
pi=4*atan2(1,1)
#
printf "Number of atoms : %d \n",n
printf "Number of electrons : %d \n",ne
printf "Number of double occupied orbitals : %d \n",npe
printf "Number of single occupied orbitals : %d \n",nue
#
# Calculate the eigenvalues
#
tEa=0
tEb=0
for (k=1;k<=n;k++) {
x=-2*cos(2*k*pi/n)
Eb[k]=x
iEb[k]=k
}
#
# Check for degenerate eigenvalues
#
for (k=1;k<=n;k++) Sim[k]=0
for (k=1;k<=n;k++) {
for (l=k+1;l<=n;l++) {
if (abs(Eb[k]-Eb[l])<0.00001) {
Sim[k]=l
Sim[l]=k
}
}
}
#
# Calculate the wavefunction coefficients
#
for (k=1;k<=n;k++) {
for (l=1;l<=n;l++) {
rc[k,l]=sqrt(1/n)*cos(2*k*(l-1)*pi/n)
ic[k,l]=sqrt(1/n)*sin(2*k*(l-1)*pi/n)
}
}
#
# Remove complex coefficients
#
for (k=1;k<=n;k++) {
sum=0
sum1=0
kk=Sim[k]
if (kk!=0 && k< kk) {
for (l=1;l<=n;l++) {
ar[l]=rc[kk,l]+rc[k,l]
ai[l]=ic[kk,l]+ic[k,l]
sr[l]=rc[kk,l]-rc[k,l]
si[l]=ic[kk,l]-ic[k,l]
sum+=(ar[l]*ar[l])
sum1+=(si[l]*si[l])
}
}
for (l=1;l<=n;l++) {
if (kk!=0 && k<kk) {
ar[l]=ar[l]/sqrt(sum)
si[l]=si[l]/sqrt(sum1)
}
}
if (kk!=0) {
for (l=1;l<=n;l++) {
rc[kk,l]=si[l]
}
for (l=1;l<=n;l++) {
rc[k,l]=ar[l]
}
}
}
printf "Orbital Energies:\n"
#
# Print the energy values
#
bubble(n,Eb,iEb)
for (k=1;k<=n;k++) {
printf " E%d=alpha %+8.3fbeta\n",k,-Eb[k]
}
#
# Calculate the total energy of the electronic system
#
npe=int(ne/2)
nue=ne-2*npe
for (i=1;i<=npe+nue;i++) {
fa=1
if (i<=npe) fa=2
tEa+=fa
tEb+=fa*Eb[n-(i-1)]
}
printf "Total Electronic Energy:\n"
printf " Epi=%8.3falpha %+8.3fbeta\n",tEa,tEb
#
# Sort the energy and wavefunctions and print them
#
printf "Wavefunctions:\n"
for (k=1;k<=n;k++) {
printf " Psi(%d)= ",k
kk=iEb[k]
for (l=1;l<=n;l++) {
printf "(%+8.3f)X(%d)",rc[kk,l],l
}
printf"\n"
}
}


Pingback: Physical Chemistry: The Simple Hückel Method (Part V) |
Pingback: Physical Chemistry: The Simple Hückel Method (Part VI): PREVIEW |