Il metodo degli orbitali molecolari di Hückel è un metodo quanto meccanico semi-empirico usato per calcolare le proprietà degli orbitali molecolari degli elettroni π in sistemi d’idrocarburici coniugati, come, per esempio, l’etilene, il benzene o il butadiene. In questa serie di articoli, ho riassunto gli aspetti principali della teoria con esempi pratici di applicazioni e programmazione del metodo.
Il metodo fu ideato da Erich Hückel nel 1930 e, successivamente ampliato e migliorato, da altri studiosi. Il metodo ha aiutato a ottenere importanti risultati teorici nell’ambito dello studio delle proprietà di molecole organiche, e dei meccanismi delle loro reazioni chimiche, in periodo precedente allo sviluppo di calcolatori elettronici. Per esempio, ha fornito le basi teoriche della regola di Hückel per l’aromaticità di (4n + 2) π sistemi planari ciclici di elettroni.
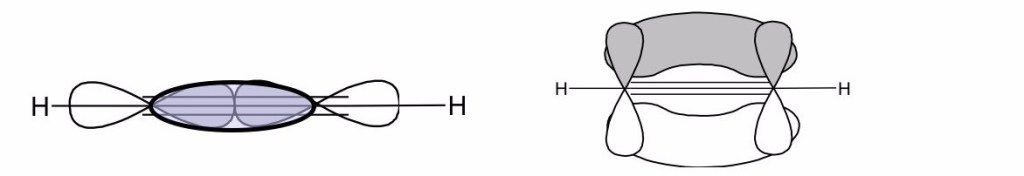
Hückel ha ipotizzato che per in molecole organiche insature gli elettroni nei legami 

Con tale ipotesi, possiamo scrivere l’energia totale del sistema molecolare come 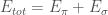
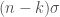


Possiamo anche supporre che la funzione d’onda molecolare dell’intero sistema possa essere approssimata da orbitali descritti come prodotto di funzioni d’onda con un elettrone
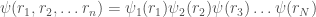
L’energia di un sistema può essere calcolata utilizzando l’operatore quantistico di Hamilton (Hamiltoniano) definito come

con
: l’energia cinetica del nucleo.
: l’energia cinetica degli elettroni.
: l’energia potenziale di attrazione protone-elettrone.
: l’energia potenziale di repulsione protone-protone.
: l’energia di repulsione elettrone-elettrone.
Nel caso di una molecola composta da atomi M, l’hamiltoniano totale può essere scritto come
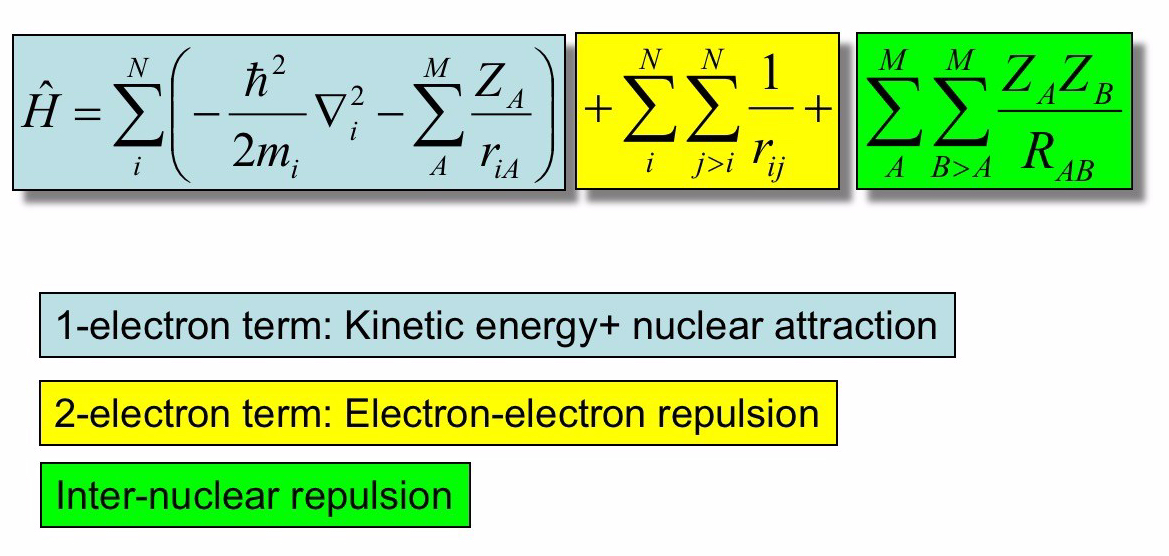
Per un sistema molecolare con elettroni π, possiamo ulteriormente distinguere questi elettroni da quelli

Per gli elettroni non d’interazione, possiamo anche supporre che l’energia totale del sistema possa essere calcolata da un operatore Hamiltoniano totale (vedi il mio blog sulla meccanica classica) può essere espressa come


e gli autostati di ciascun elettrone possono essere calcolati dalle equazioni di Schrödingen

Come ulteriore ipotesi, la funzione d’onda degli elettroni 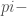
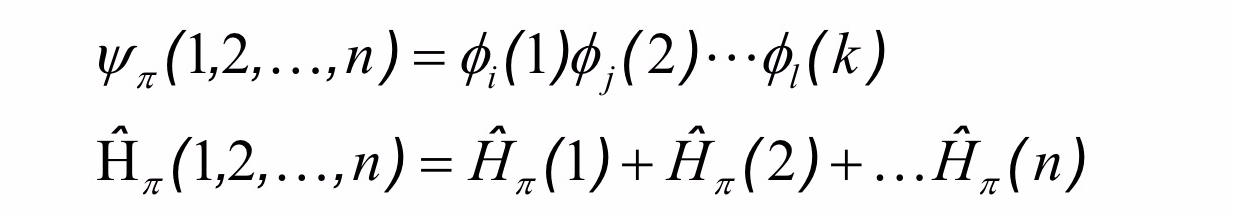
Usando queste approssimazioni, l’energia elettronica totale degli elettroni 
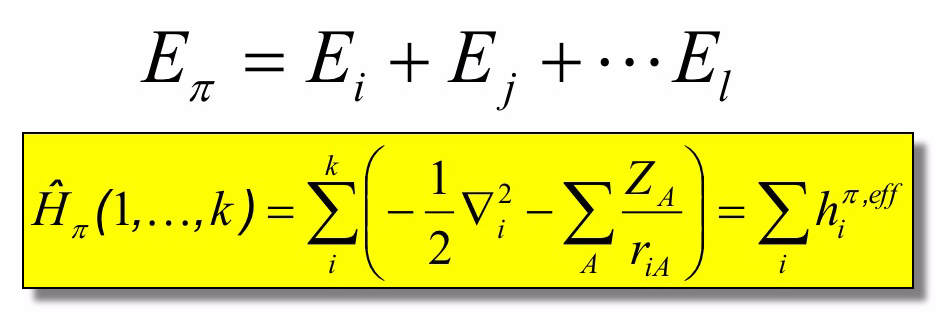
Ora considereremo un semplice sistema molecolare (la molecola dell’allile) per mostrare come assegnare gli orbitali atomici degli atomi per costruire il determinante di Hückel. Una molecola allilica ha la formula strutturale 


Consideriamo i tre orbitali 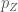
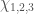
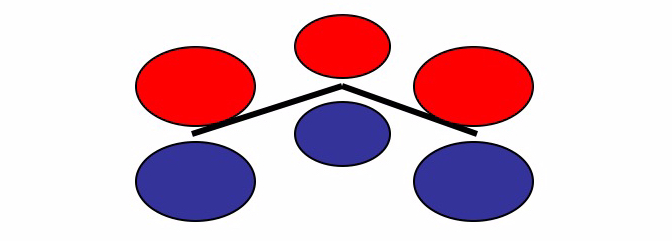
L’orbitale molecolare totale può essere considerato come una combinazione lineare di questi orbitali atomici.
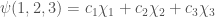
Per trovare l’energia del sistema molecolare, utilizzeremo il metodo basato sul risultato del teorema variazionale.
Il teorema di variazione afferma che dato un sistema con un operatore hamiltoniano 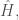
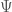

dove 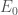
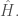
Il principio variazionale ci consente di calcolare un limite superiore per l’energia dello stato fondamentale trovando la funzione d’onda di prova 

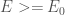
Il metodo variazionale lineare
Utilizzeremo il cosiddetto metodo della variazione lineare (o metodo di Rayleigh-Ritz) in cui la funzione di variazione lineare è una combinazione lineare di 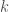
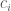
Inserendo la funzione
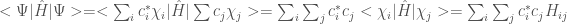
dove 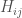
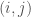

dove 
Possiamo quindi scrivere

o nella forma riorganizzata come

Ora dobbiamo trovare il valore dei coefficienti 
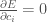
per tutte le funzioni i.
Differenziando entrambi i membri della (1) e utilizzando le proprietà di
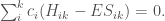
Abbiamo quindi un insieme di k equazioni omogenee simultanee (equazioni secolari) in k incognite. Nel caso della molecola d’allile, il sistema è il seguente
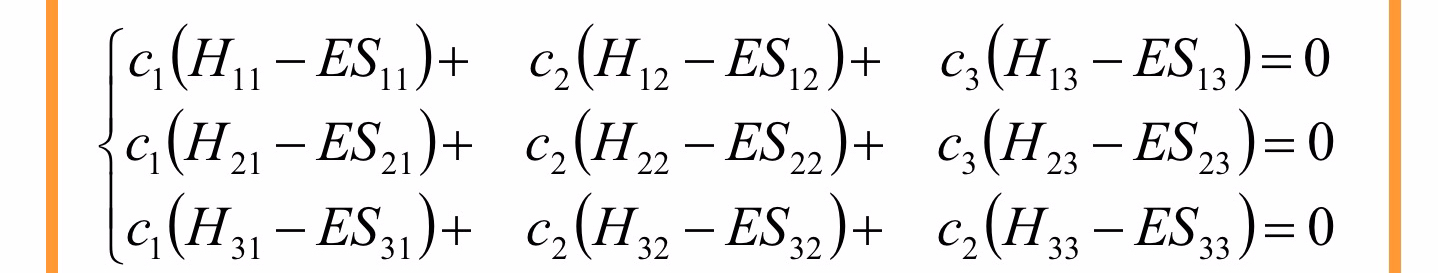
Per una soluzione non banale (cioè 
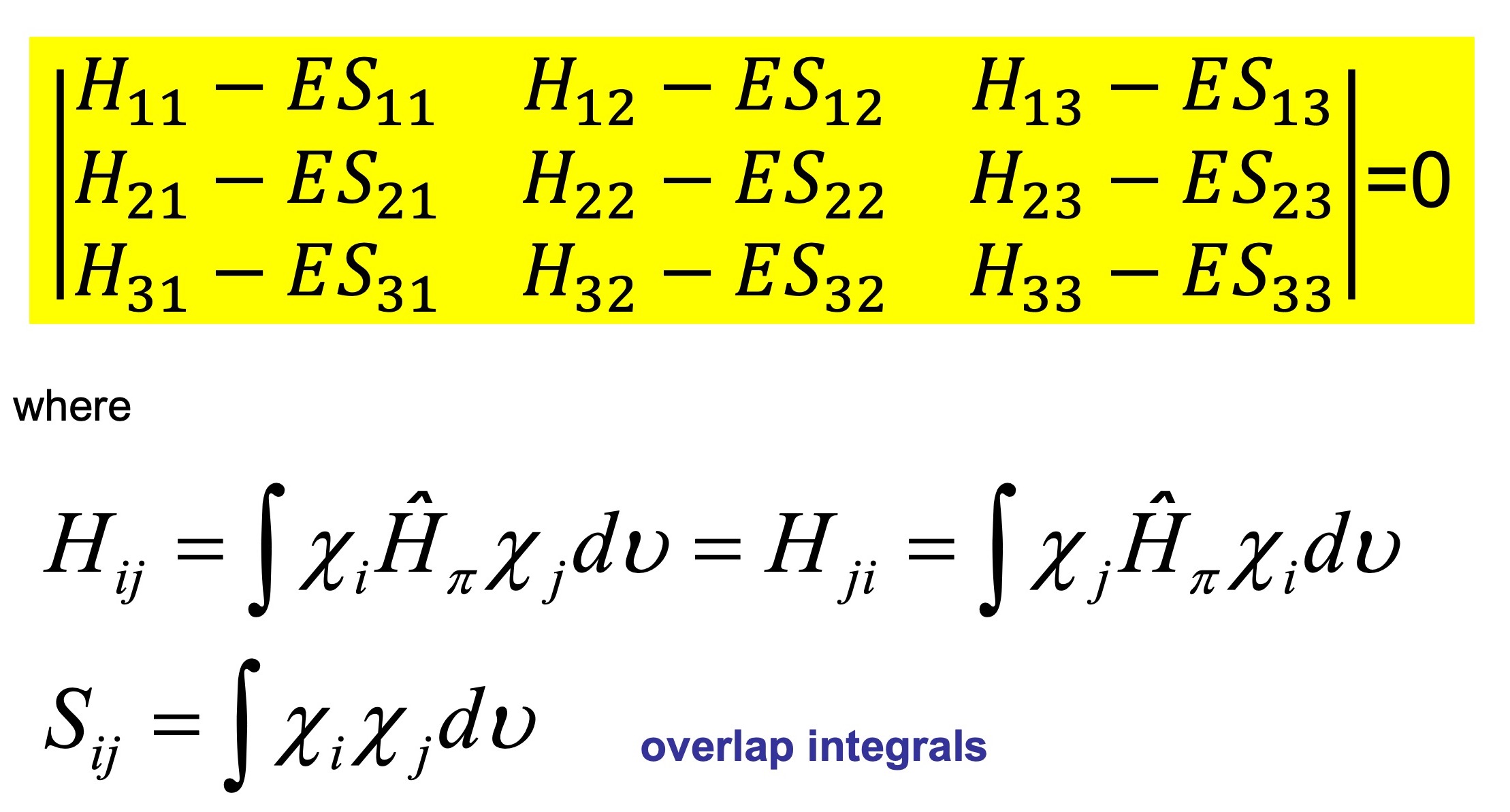
L’integrale 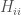
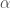
L’integrale 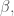
Pertanto possiamo impostare i valori dell’integrale

e poiché gli orbitali sono normalizzati, gli integrali di sovrapposizione sono dati dalla espressione
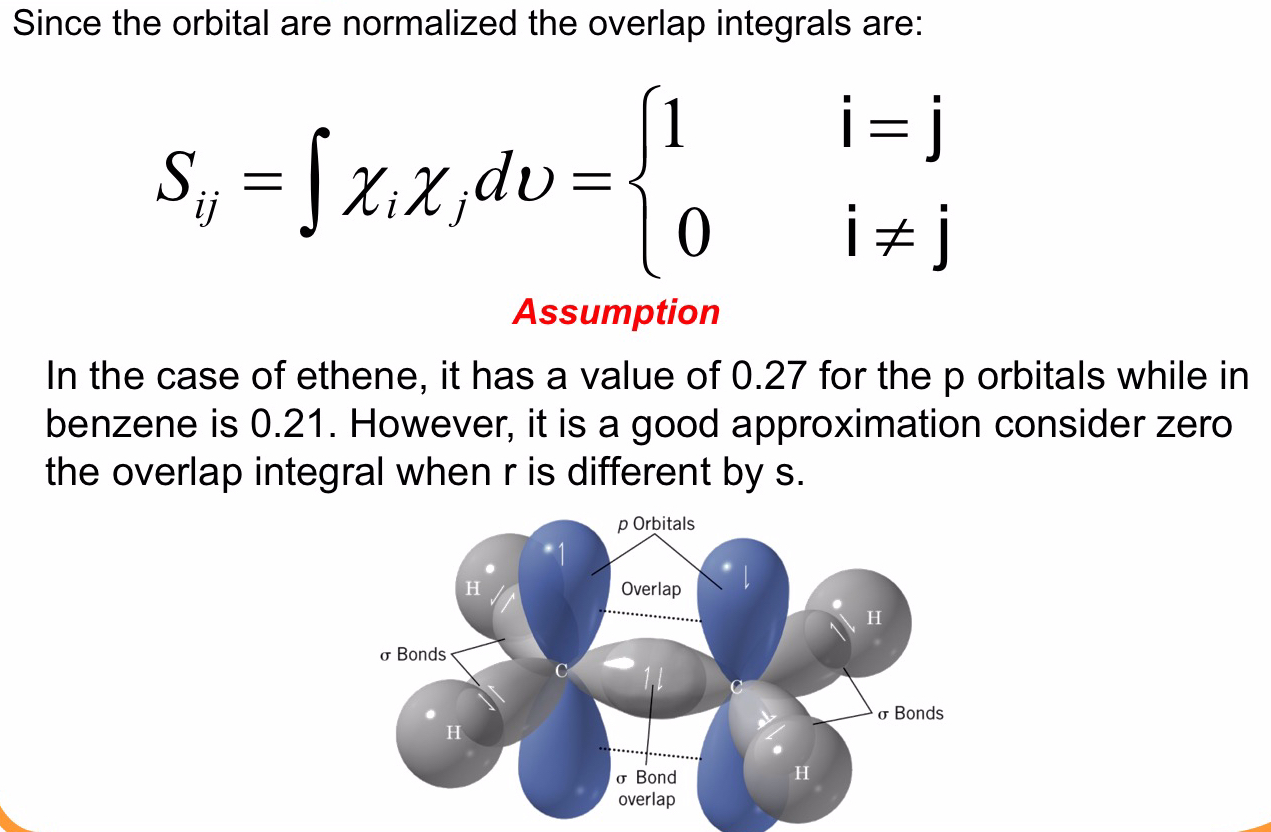
Con queste semplificazioni diventa il determinante di Hückel per la molecola dell’ allile è il seguente


Nella figura che segue, sono mostrati altri esempi di determinante di Hückel per semplici molecole insature
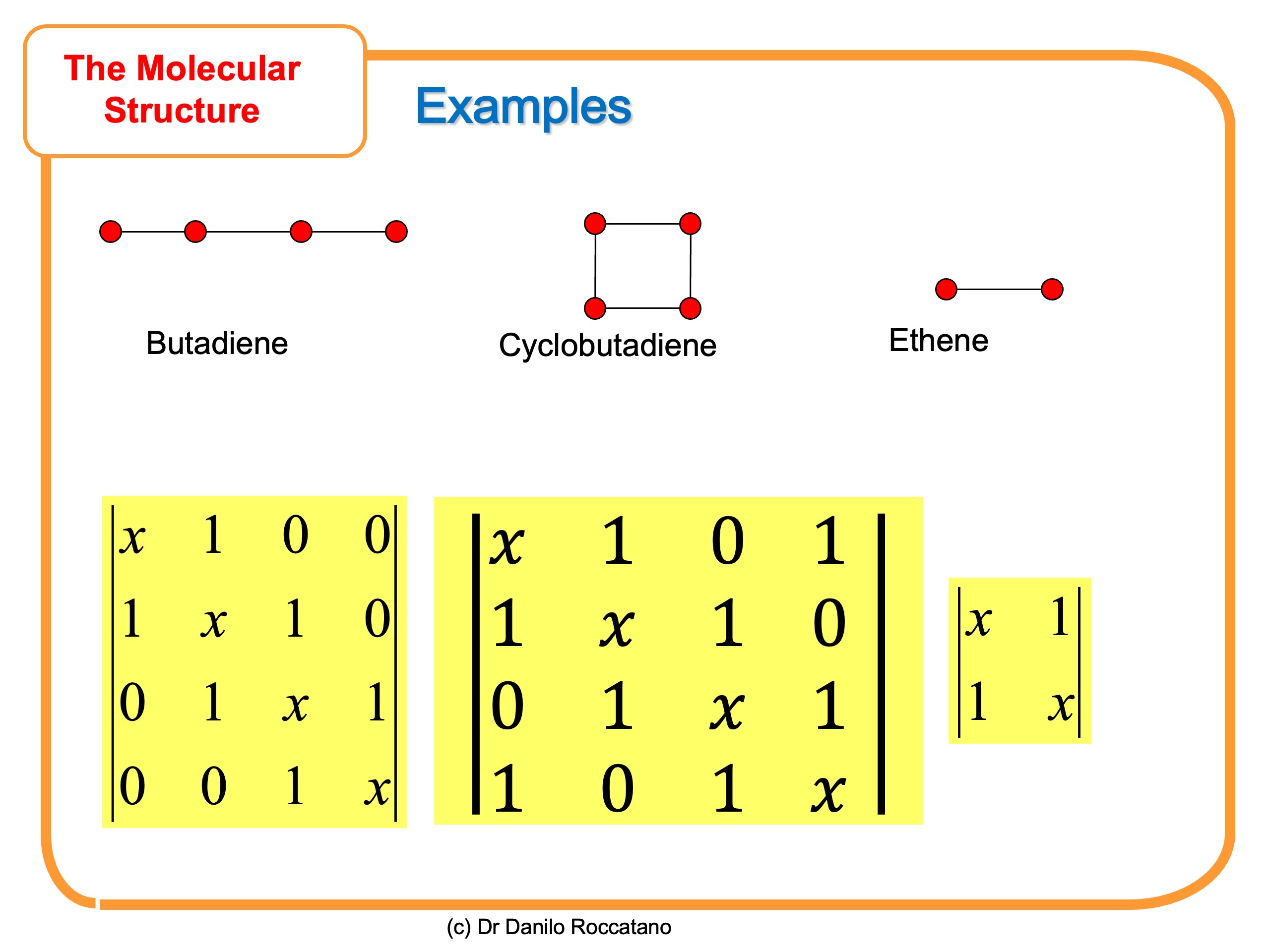
Nella seconda parte di questo articolo, impareremo come risolvere l’equazione caratteristica del determinate secolare per trovare l’energia degli orbitali molecolari.
Se hai trovato utile questo articolo, condividilo con i tuoi amici e non dimenticare di aggiungere un Like e d’iscriviti per ricevere aggiornamenti!
REFERENCES
- J.P. Lowe. Quantum Chemistry. 1993, Academic Press.

