Le cariche parziali da usare nel campo di forze si ottengono generalmente da calcoli quantomeccanici. Nel caso di molecole rigide, il calcolo delle cariche è abbastanza semplice. Nel caso di molecole flessibili, occorre valutare quanto le diverse conformazioni influenzano la distribuzione di carica e, quindi, stimare la carica parziale come media pesata tra i vari conformeri. Il calcolo QM fornisce le cosidette cariche di Mulliken. Questo tipo di cariche possono portare ad una elevata inaccuratezza nel riprodurre proprietà chimico-fisiche di piccole molecole. Per evitare questo inconveniente sono state introdotte varie procedure per ottenere delle cariche parziali che tengano conto della diversa capacitá dei singoli atomi di accomodare una diversa distribuzione di carica. Queste procedure vanno sotto il nome di metodi di Electrostatic potential fitting tra cui i più usati solo il RESP e il CHELPG. Vediamo come questi metodi funzionano.
Il potenziale elettrostatico 
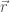

dove 



ovvero, la somma dei minimi quadrati della differenza tra il potenziale quantomeccanico e quello calcolato da queste cariche in un certo numero di punti 



dove 

derivando 

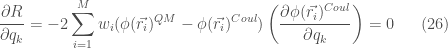
che può essere scritta come un set di M equazioni
![\displaystyle \sum_{i=1}^{M} w_i\left(\phi (\vec{r_i})^{QM}-\frac{Z}{r_{iN}}\right)\left(\frac{1}{r_{ik}}-\frac{1}{r_{iN}}\right) = \sum_{j=1}^{N-1}\left[\sum_{i=1}^{M} w_i \left(\frac{1}{r_{ik}}-\frac{1}{r_{iN}}\right) \left(\frac{1}{r_{ij}}-\frac{1}{r_{iN}}\right)\right] \frac{q_j}{4\pi \epsilon_0} \ \ \ \ \ (27)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Csum_%7Bi%3D1%7D%5E%7BM%7D+w_i%5Cleft%28%5Cphi+%28%5Cvec%7Br_i%7D%29%5E%7BQM%7D-%5Cfrac%7BZ%7D%7Br_%7BiN%7D%7D%5Cright%29%5Cleft%28%5Cfrac%7B1%7D%7Br_%7Bik%7D%7D-%5Cfrac%7B1%7D%7Br_%7BiN%7D%7D%5Cright%29+%3D+%5Csum_%7Bj%3D1%7D%5E%7BN-1%7D%5Cleft%5B%5Csum_%7Bi%3D1%7D%5E%7BM%7D+w_i+%5Cleft%28%5Cfrac%7B1%7D%7Br_%7Bik%7D%7D-%5Cfrac%7B1%7D%7Br_%7BiN%7D%7D%5Cright%29+%5Cleft%28%5Cfrac%7B1%7D%7Br_%7Bij%7D%7D-%5Cfrac%7B1%7D%7Br_%7BiN%7D%7D%5Cright%29%5Cright%5D+%5Cfrac%7Bq_j%7D%7B4%5Cpi+%5Cepsilon_0%7D+%5C+%5C+%5C+%5C+%5C+%2827%29&bg=ffffff&fg=444444&s=0&c=20201002)
o, in forma matriciale 
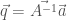
- CHELPG: sviluppato da Breneman e Wiberg nel 1990. In questo metodo, la molecola viene posta in una reticolo cubico di punti (con spaziatura compresa tra 0.3 e 0.8
). Tutti i punti che si trovano entro il raggio di van der waals e
al di fuori di questo vengono scartati, e i punti rimanenti usati per il calcolo.
- RESP: sviluppato dal gruppo di P. Kolmann per calcolare le cariche parziali da usare nel campo di forza AMBER. In questo caso, i punti vengono presi da superfici molecolari costruite usando raggi di van der waals progressivamente crescenti, e imponendo dei vincoli su particolari atomi (per esempio carboni localizzati all’interno della molecola) che potrebbero altrimenti assumere cariche eccessivamente alte. A differenza del CHELPG, le cariche ottenute con questo metodo sono meno sensibili alla conformazione molecolare.
Per effettuare il calcolo delle cariche parziali si può ricorrere ai programmi per calcoli di QM, GAUSSIAN o GAMESS/US (quest’ultimo è un programma liberamente distribuito). Per il calcolo è necessario disporre di coordinate iniziali della molecola. Queste possono essere ottenute dal data base di strutture (come il Cambridge Structural Database) oppure costruite usando programmi di modellistica molecolare. Un programma molto diffuso di questo tipo è MOLDEN che può essere liberamento scaricato dal sito: http://www.cmbi.ru.nl/molden
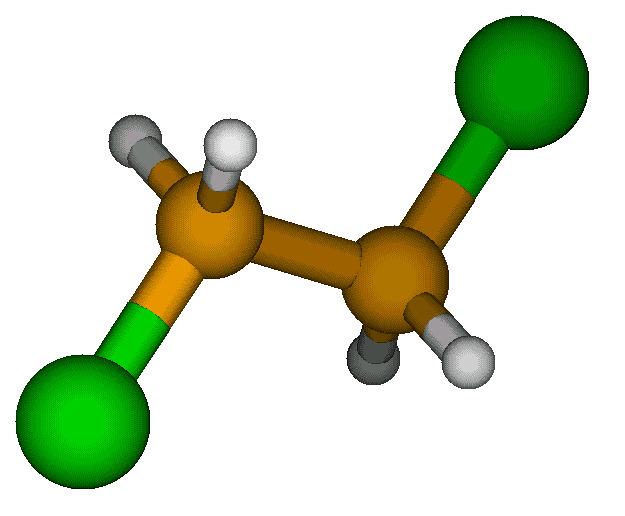
Usando questo programma è possibile costruire la struttura iniziale della molecola e salvare su file le coordinate nel formato GAUSSIAN o GAMESS/US, per il calcolo delle cariche. Come esempio di calcolo delle cariche viene mostrato il contenuto del file di input per GAUSSIAN per al calcolo a punto singolo delle conformazioni syn, gauge e anti del 1,2,dicloroetano.
%Chk=DCLETANO # RHF/6-31G(d) FOpt Test Pop=(CHelpG,dipole) Calcolo delle cariche con il metodo di CHELPG 0 1 c cl 1 clc2 c 1 cc3 2 cccl3 h 1 hc4 2 hccl4 3 dih4 h 1 hc5 2 hccl5 3 dih5 cl 3 clc6 1 clcc6 2 60. h 3 hc7 1 hcc7 6 dih7 h 3 hc8 1 hcc8 6 dih8 clc2 1.750000 cc3 1.540000 cccl3 109.471 hc4 1.089000 hccl4 109.471 dih4 120.000 hc5 1.089000 hccl5 109.471 dih5 -120.000 clc6 1.750000 clcc6 109.471 hc7 1.089000 hcc7 109.471 dih7 120.000 hc8 1.089000 hcc8 109.471 dih8 240.000 dih6 0.0
Come si può vedere dalla prima linea, i programmi effettuano dapprima una minimizzazione delle coordinate, e una volta che si è raggiunta la convergenza viene effettuato un calcolo ESP usando il metodo CHELPG.
Nella tabella I, sono riportati i valori delle cariche ottenute per le varie configurazioni e il relativo momento di dipolo.
6.2. Calcolo di un profilo di energia potenziale per rotazione di un legame.
Quando in una molecola sono presenti vari angoli diedri, può essere utile calcolare il profilo dell’energia conformazionale ottenuta dalla rotazione intorno a questi. Questo profilo può essere usato per modellare l’energia potenziale classica dovuta alla rotazione intorno al suddetto diedro, usando il potenziale di Rycheman-Belleman ed escudendo le interazioni di non-legame intramolecolari tra gli atomi coinvolti.
In GAUSSIAN è possibile calcolare automaticamente un profilo di energia conformazionale. Nell’esempio che segue viene mostrato come ottenere, il profilo di energia conformazionale per la molecola di 1,2-dicloroetano. Di seguito viene mostrato il file di input per GAUSSIAN per il calcolo del profilo di energia.
%Chk=DCLETANO # RHF/6-311G(D) Opt=Z-matrix NoSymm Test Relaxed PES scan 0 1 c cl 1 clc2 . . . dih6 0. 18 10.
Nell’ultima riga viene indicato l’angolo diedro Cl-C-C-Cl (dih6) interessato alla rotazione. La scansione viene effettuata ruotando 18 volte di 10 gradi l’angolo diedro. Per estrarre dal file di uscita il profilo di energia in funzione dell’angolo si può usare lo script in awk riportato alla fine di questo articolo.
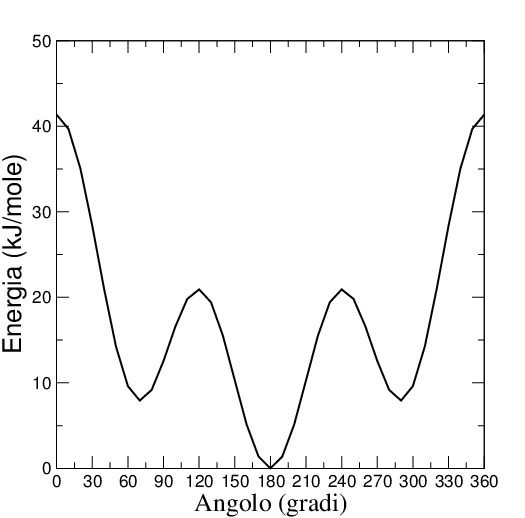
Nella Figura 2, viene riportato il grafico del profilo di energia ottenuto dalla scansione dell’angolo diedro.
#! /usr/bin/awk -f
BEGIN {
# Incremental step of the scansion
step = 10.
# Conversion factor Hartree --> Kcal/mol
conv = 627.5095
cc = 0
count = 0
}
$1 == "EIGENVALUES" { for (i=3;i<=NF;i++) {
k=split($i,a,"-")
for (ii=1;ii<=k;ii++) {
if (a[ii] != "") {
val = -a[ii]
en[cc] = val
if (val < min) {min = val}
count+=step
cc++
}
}
}
}
END {
count = 0
for (ii=0;ii<cc;ii++) {
printf "% 4.1f %8.3f\n", count, (en[ii]-min)*conv
count+=step
}
}

