Then from these forces, by other propositions which are also mathematical, I deduce the motions of the planets, the comets, the moon, and the sea. I wish we could derive the rest of the phenomena of Nature by the same kind of reasoning from mechanical principles, for I am induced by many reasons to suspect that they may all depend upon certain forces by which the particles of bodies, by some causes hitherto unknown, are either mutually impelled towards one another, and cohere in regular figures, or are repelled and recede from one another.
Isaac Newton. Philosophiae Naturalis Principia Mathematica. London, 1686.
La funzione di potenziale usata nella DM per descrivere le interazioni tra gli atomi del sistema è chiamata campo di forze. Si tratta di funzioni analitiche che dipendono dalle coordinate atomiche 

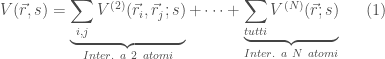
Un esempio di campo di forza, utilizzato in molti programmi per simulazioni di DM, è il seguente:

Equazione 1: Tipica espressione di un force field usato per simulazioni di DM.
1. Interazioni di legame
Sono descritte da termini a 2, 3 e 4 corpi, generalmente armonici, che contengono costanti ottenute da dati sperimentali cristallografici e spettroscopici.
1.1. Vibrazioni di legame
Il primo termine (
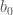
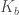


Figura 1: Curve di potenziale per le vibrazione di legame: armonico (a destra); Morse (a sinistra).
In alcuni campi di forze si utilizza la funzione di Morse:
![\displaystyle V_M(b)=\sum_{legami} D_e \left\{ e^{[ - k_b(b-b_0)]} -1 \right\}^2 \ \ \ \ \ (3)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+V_M%28b%29%3D%5Csum_%7Blegami%7D+D_e+%5Cleft%5C%7B+e%5E%7B%5B+-+k_b%28b-b_0%29%5D%7D+-1+%5Cright%5C%7D%5E2+%5C+%5C+%5C+%5C+%5C+%283%29&bg=ffffff&fg=444444&s=0&c=20201002)
dove il parametro 

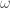
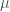

1.2. Vibrazione degli angoli di legame
Il secondo termine nella Equazione 1 si riferisce all’energia dovuta alla deformazione dell’angolo di valenza ( interazione a tre corpi). Anche questo ha la forma di potenziale armonico dove 


Figure 2: Potenziale armonico di vibrazione dell’angolo di legame.
Esiste un gran numero di varianti dell’equazione 1. Alcuni campi di forza prevedono termini misti tipo ![{K_{b\theta}[b-b_0][\theta-\theta_0]}](https://s0.wp.com/latex.php?latex=%7BK_%7Bb%5Ctheta%7D%5Bb-b_0%5D%5B%5Ctheta-%5Ctheta_0%5D%7D&bg=ffffff&fg=444444&s=0&c=20201002)
1.3. Interazioni a 4 corpi
Due termini sono utilizzati per descrivere le interazioni a quattro corpi.
1.3.1 Gli angoli diedri impropri.
Il primo termine è usato per descrivere i cosiddetti angoli diedri impropri, che sono introdotti per correggere due artefatti: la deformazione delle geometrie tetraedriche e planari. La deformazione delle geometrie tetraedriche è dovuta al fatto che nella maggior parte dei programmi di DM, gli idrogeni apolari (cioè quelli legati agli atomi di carbonio) non vengono trattati esplicitamente, ma viene incluso il loro effetto modificando le funzioni di potenziale degli atomi a cui sono legati. In tal modo un carbonio chirale, legato a un idrogeno, viene simulato con solo tre legami; questo comporta che durante la simulazione si possono avere delle inversioni di chiralità rendendo, quindi, necessario l’uso di questo ulteriore termine di potenziale per vincolare la geometria del sito. L’ angolo diedro improprio, A-X-Y-B, è definito come l’angolo tra il piano che passa per gli atomi A, X e Y e il piano passante per gli atomi X, Y e B.
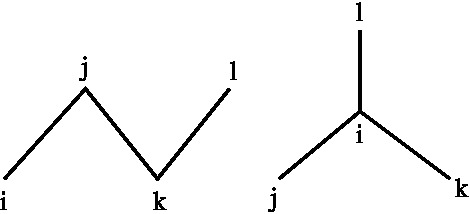
Figure 3: Definizione dei diedri impropri.
1.3.2 Gli angoli diedri propri
L’altro termine per le interazioni a quattro atomi è quello relativo ai normali angoli diedri ed è una funzione sinusoidale, 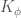
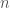


Figure 4: Andamento della funzione di potenziale di rotazione intorno a un diedro proprio.
2. Parametri per le interazioni di legame
Per determinare i parametri per i campi di forza si possono seguire due strade. Il metodo più elegante è tarare questi parametri con i risultati di calcoli quantistici ab-initio su piccoli aggregati molecolari. I campi di forza ottenuti in tal modo, molto spesso, non danno risultati soddisfacenti date le molte approssimazioni che devono essere fatte in questo tipo di procedura. In alternativa è possibile tarare i parametri del campo di forza con dati sperimentali quali: strutture cristallografiche, energie e dinamiche di reticolo, dati spettroscopici, proprietà di liquidi tipo densità e entalpia di vaporizzazione, energia libera di solvatazione, dati di RMN, ecc [Jorgensen83, Lifson83, Giglio69, DiNola70]. In questo modo viene garantita una maggiore riproducibilità delle proprietà macroscopiche di massa di liquidi, in quanto i parametri sperimentali tengono conto implicitamente dell’effetto del campo medio delle molecole del solvente.
2.1 Interazioni di non legame
L’ultimo termine nella equazione 1 descrive le interazioni tra coppie di atomi non legati. Queste sono le interazioni di van der Waals e Coulombiane tra gli atomi 
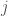



2.1.1. Interazioni di van der Waals
Una delle piú utilizzate espressioni per la descrizione del potenziale d’interazione di non legame è il potenziale di Lennard-Jones. Il potenziale di Lennard-Jones è formato da due termini che descrivono una interazione di tipo repulsivo e una di tipo attrattivo:
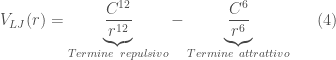


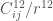
![{b\exp [-ar]}](https://s0.wp.com/latex.php?latex=%7Bb%5Cexp+%5B-ar%5D%7D&bg=ffffff&fg=444444&s=0&c=20201002)



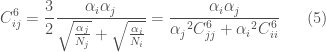
in cui 

I valori di 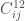
NOTA: L’equazione di stato di un gas reale può essere espressa nella forma del viriale
dove 

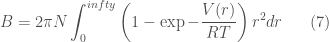
dove r è la distanza da una molecola scelta come origine. Sostituendo in V(r) l’espressione della equazione di Lennard-Jones, si ottengono espressioni che permettono di determinare i parametri 
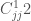
Una forma molto usata del potenziale di Lennard-Jones, sostituisce a 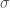

![\displaystyle V_{LJ} (r)= 4 \epsilon \left[\left(\frac{\sigma}{r}\right)^{12} - \left(\frac{\sigma}{r}\right)^{6}\right] \ \ \ \ \ (8)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+V_%7BLJ%7D+%28r%29%3D+4+%5Cepsilon+%5Cleft%5B%5Cleft%28%5Cfrac%7B%5Csigma%7D%7Br%7D%5Cright%29%5E%7B12%7D+-+%5Cleft%28%5Cfrac%7B%5Csigma%7D%7Br%7D%5Cright%29%5E%7B6%7D%5Cright%5D+%5C+%5C+%5C+%5C+%5C+%288%29&bg=ffffff&fg=444444&s=0&c=20201002)
I valori di

Figura 5: Curva di Lennard-Jones con i parametri.
Esistono diversi tipi di regole di combinazione, in una delle più usate i nuovi parametri si ottengono in questo modo:


Le interazioni di non legame tra atomi legati covalentemente o separati da due legami non vengono generalmente calcolate. Nel caso, invece, di interazioni tra atomi separati da tre legami i parametri delle funzioni di LJ sono opportunamente ridotti per evitare le forti repulsioni che si avrebbero a distanze corte. I parametri 

2.1.2. Interazioni elettrostatiche
L’interazione elettrostatica viene generalmente descritta attraverso un termine coulombiano. Ad ogni atomo del sistema viene quindi attribuita una carica atomica parziale che vengono calcolate con metodi quantomeccanici (vedi appendice). La scelta della costante dielettrica relativa 
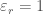


dove N è il numero di atomi del sistema, 

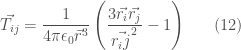
le precedenti equazioni danno luogo a un sistema di 3N equazioni in 3N incognite (ciascuna delle componenti x, y, z di ogni atomo) che può essere risolto algebricamente o iterativamente. Un altro modo elegante di introdurre la polarizzabilià nel sistema è quello di usare delle cariche puntiformi, localizzate su ciascun atomo e legate da tra loro da oscillatori di opportuna costante di forza (oscillatori di Drude), la loro deformazione in seguito alla interazioni con altri atomi produce una separazione delle cariche e quindi un dipolo indotto
2.1.3. Il legame a idrogeno
Un’ altra importante interazione molecolare è il legame idrogeno. Il termine legame idrogeno viene usato per indicare quasi tutte quelle situazioni in cui le distanze internucleari, 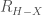
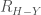



che viene usato per descrivere l’interazione tra il donatore dell’atomo di idrogeno e l’atomo accettore. In altri potenziali, la precedente funzione viene moltiplicata per un termine che tiene conto dell’orientamento tra gli atomi coinvolti.
3. I termini speciali
All’equazione 1 possono essere aggiunti, alcuni termini speciali che, vincolando alcuni gradi di libertà del sistema limitano, lo spazio conformazionale esplorato sulla base dei dati strutturali ottenuti sperimentalmente. Questi termini costituiscono la base per l’ affinamento delle strutture molecolari ottenute da misure di risonanza magnetica nucleare (RMN) o di diffrazione dei raggi X.
3.1. Potenziali di vincolo delle posizioni
Se nel corso di una simulazione si vogliono tenere bloccati le posizioni nello spazio di alcuni atomi del sistema, si può ricorrere all’uso di potenziali armonici del tipo:

dove N indica il numero di atomi a cui applicare il potenziale di vincolo, 

3.2. Potenziali di vincolo delle distanze
La spettroscopia RMN permette di ottenere informazioni sulle distanze interprotoniche (tramite l’effetto Overhauser interprotonico) e su angoli diedri (dalle costanti di accoppiamento J) e sugli spostamenti chimici.
Le intensità NOE (Nuclear Overhauser Effect) possono essere convertite in un insieme di limiti superiori 
![\displaystyle V(\vec{r})=\frac{1}{2} \sum_{NOE}\left[MAX(0,<r_{ij}^{-1/3}>-r_{ij}^{ub})\right]^2 \ \ \ \ \ (15)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+V%28%5Cvec%7Br%7D%29%3D%5Cfrac%7B1%7D%7B2%7D+%5Csum_%7BNOE%7D%5Cleft%5BMAX%280%2C%3Cr_%7Bij%7D%5E%7B-1%2F3%7D%3E-r_%7Bij%7D%5E%7Bub%7D%29%5Cright%5D%5E2+%5C+%5C+%5C+%5C+%5C+%2815%29&bg=ffffff&fg=444444&s=0&c=20201002)
dove la funzione MAX fornisce il valore del maggiore tra i due argomenti.
I valori delle costanti di accoppiamento 
![\displaystyle V_{J}(\vec{r})= \frac {1}{2} K_{J} \sum_{\phi_i} \left[ <J(\phi_i(\vec{r})> - J_i^{spe}\right]^2 \ \ \ \ \ (16)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+V_%7BJ%7D%28%5Cvec%7Br%7D%29%3D+%5Cfrac+%7B1%7D%7B2%7D+K_%7BJ%7D+%5Csum_%7B%5Cphi_i%7D+%5Cleft%5B+%3CJ%28%5Cphi_i%28%5Cvec%7Br%7D%29%3E+-+J_i%5E%7Bspe%7D%5Cright%5D%5E2+%5C+%5C+%5C+%5C+%5C+%2816%29&bg=ffffff&fg=444444&s=0&c=20201002)
Le costanti di accoppiamento 
Infine, gli spostamenti chimici 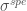
![\displaystyle V_{\sigma}(\vec{r})= \frac {1}{2} K_{\sigma} \sum_{i} \left[ <\sigma_i(\vec{r})> - \sigma_i^{spe}\right]^2 \ \ \ \ \ (17)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+V_%7B%5Csigma%7D%28%5Cvec%7Br%7D%29%3D+%5Cfrac+%7B1%7D%7B2%7D+K_%7B%5Csigma%7D+%5Csum_%7Bi%7D+%5Cleft%5B+%3C%5Csigma_i%28%5Cvec%7Br%7D%29%3E+-+%5Csigma_i%5E%7Bspe%7D%5Cright%5D%5E2+%5C+%5C+%5C+%5C+%5C+%2817%29&bg=ffffff&fg=444444&s=0&c=20201002)
dove la sommatoria è effettuata su tutte le risonanze osservate. La media 
3.3. Potenziali di vincolo sui fattori di struttura cristallografici
Nel caso in cui si disponga di dati strutturali ottenuti da misure di diffrazione dei raggi X (ampiezza dei fattori di struttura 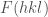

![\displaystyle V_{X}= \frac{1}{2}K_{X} \sum_{hkl}\left[F_{calc}(hkl)-F_{obs}(hkl)\right]^2 \ \ \ \ \ (18)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+V_%7BX%7D%3D+%5Cfrac%7B1%7D%7B2%7DK_%7BX%7D+%5Csum_%7Bhkl%7D%5Cleft%5BF_%7Bcalc%7D%28hkl%29-F_%7Bobs%7D%28hkl%29%5Cright%5D%5E2+%5C+%5C+%5C+%5C+%5C+%2818%29&bg=ffffff&fg=444444&s=0&c=20201002)
dove 
L’uso dei valori medi nelle precedenti funzioni di vincolo le rende non conservative (poichè diventano dipendenti dal tempo), questo rende necessario dissipare il calore generato accoppiando il sistema a un bagno termico esterno [Torda90].
4. Campi di forze e librerie di parametri
Esistono vari tipi di campi di forza che sono stati sviluppati da diversi gruppi di ricerca in tutto il mondo. Questi campi di forza sono costituiti da librerie di parametri ottimizzati per varie classi di molecole. Le librerie di parametri più comuni sono quelle ottimizzate per lo studio di proteine o acidi nucleici. Le librerie più usate sono le seguenti:
- GROMOS: il campo di forza GROningen MOlecular Simulations è stato realizzato all’Università di Groningen da H.J.C. Berendsen e W.F. van Gunsteren. Nella sua attuale versione (GROMOS96) [vanGunsteren98], viene utilizzato nel pacchetto di porogrammi per DM GROMOS96 e Gromacs.
- OPLS: L’ Optimized Potential for Liquid Simulations è stato sviluppato da W.L. Jorgensen e J. Tirado-Rives~\cite{Jorgensen88} e inizialmente utilizzato per simulazioni Monte Carlo, è diventata una delle librerie più estese e dettagliate. Esistono centinaia di diversi tipi atomici ottimizzati per differenti classi di molecole organiche. Le caratteristiche di questa libreria sono:
- Le cariche iniziali per la procedura di ottimizzazione dei parametri sono ottenute da calcoli quantomeccanici (set di base 6-31G* o 6-31+G* per anioni).
- I parametri iniziali della funzione di Lennard-Jones sono gli stessi quelli usati per gli stessi tipi di atomi in molecole simili dello stesso tipo.
- Un primo tentativo per migliorare le cariche parziali e i parametri di LJ, viene fatto modellando queste funzioni sui risultati energetici e strutturali ottenuti da calcoli QM su complessi acqua-molecola usando come set di base il 6-31G* o il 6-31+G*.
- Per molecole neutre i parametri sono ulteriormente migliorati attraverso simulazioni del liquido puro. La densità, l’entalpia di vaporizzazione, la capacità termica a pressione costante e la costante di diffusione calcolate sono confrontate con i valori sperimentali. I valori delle cariche e dei parametri di Lennard-Jones vengono quindi modificati fino a riprodurre i dati in fase gassosa e nel liquido.
- I parametri per molecole cariche sono verificati calcolando le energie libere di solvatazione e di legame.
- Da notare che le funzioni per la descrizione della vibrazione di angoli e legami non sono proprie del OPLS poichè questo campo di forze venne inizialmente utilizzato per simulazioni Monte Carlo che non consentono il cambiamento di questi parametri. Per usare questo campo di forze in programmi di DM si utilizzano come funzioni di legame quelle di AMBER.
- CHARMM: Il campo di forza CHARMM (Chemistry at HARvard, Macromolecular mechanics) e l’omonimo programma, sono stati sviluppati da M. Karplus e B. R. Brooks [Brooks83, MacKerell95] alla Univerità di Harvard. Le caratteristiche di questo campo di forze possono essere riassunte nei seguenti punti:
- Le cariche parziali vengono determinate in modo che il calore di sublimazione, i momenti di dipolo del composto nonchè le energie d’interazioni, le geometrie in fase gassosa di complessi molecola-acqua, vengano riprodotte. Le energie di interazione ottenute dal calcolo QM al livello 6-31G*, vengono scalate di un fattore 1.16, cioè dello stesso fattore che esiste tra le energie di interazione delle molecole di acqua nel modello TIP3P e quelle ottenute dal calcolo QM allo stesso livello.
- Le costanti di forza per le vibrazioni di legami, angoli e diedri sono tali da riprodurre le proprità geometriche e vibrazionali del composto, ottenute da strutture cristallografiche, spettri IR e Raman e da calcoli QM nel vuoto.
- AMBER: L’ Assisted Model Building using Energy Refinement [Weiner86, Cornell95] è il campo di forza presente nell’omonimo pacchetto di programmi per DM sviluppato da P. Kollman, all’ UCSF. Le caratteristiche di questo campo di forza possono essere riassunte nei seguenti punti:
- Le cariche originarie furono ottenute modellando le energie di interazione ottenute dal calcolo QM a livello STO-3G su composti modello.
- In versioni più recenti del campo di forza, le cariche sono state migliorate attraverso il calcolo della densità elettronica a livello 6-31G* e il fitting di questa con il metodo RESP (vedi appendice).
- Come parametri di Lennard-Jones si usano quelli del campo di forza OPLS.
- Le costanti di forza per le vibrazioni di legami e angoli vengono scelti in modo da riprodurre le frequenze sperimentali e teoriche dei modi normali di composti modello.
- I lone pairs presenti sugli atomi di zolfo sono esplicitamente trattati.
- CVFF: Il Consistent-Valence Force Field è stato proposto da Dauber-Osguthorpe [Dauber-Osguthorpe88] e viene usato nel programma Discover. Nella equazione (19), viene riportato la funzione di potenziale che descrive questo campo di forza. Una grande attenzione è stata data a creare un campo di forze che fosse in grado di riprodurre le caratteristiche vibrazionali di molecole di interesse biochimico. Questo è evidente dalla presenza nel potenziale, di numerosi termini di vibrazione accoppiati (legame-legame, legame-angolo, angolo-angolo, angolo-diedro, diedro-diedro).
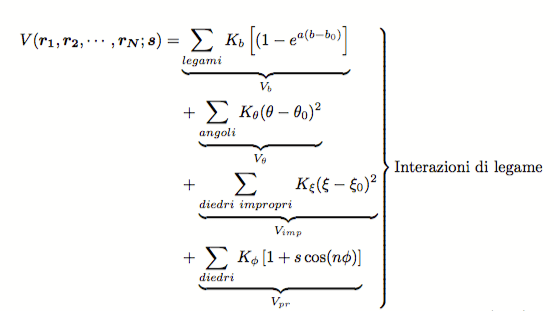
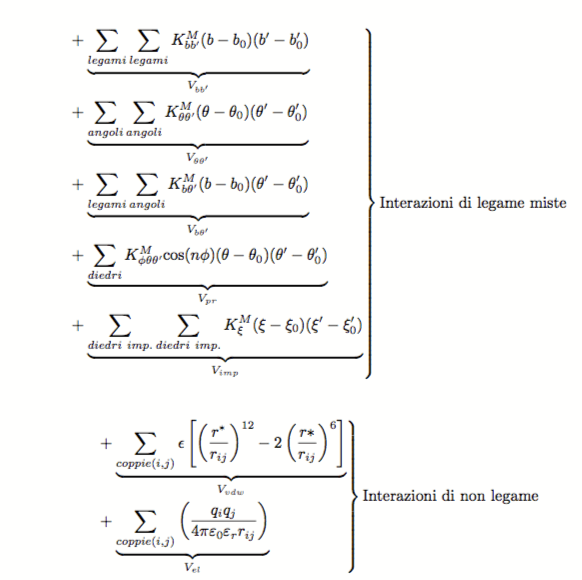
(19)
5. Ottimizzazione della geometria molecolare
Il campo di forza, descritto nei precedenti paragrafi, viene usato in meccanica molecolare per costruire la superficie di energia conformazionale della molecola e quindi ricercare quelle conformazioni che sono energeticamente più stabili, cioè localizzate in punti di minimo. Questa procedura, come vedremo, è molto importante nella fase di partenza delle simulazioni di DM per evitare che configurazioni molecolari troppo stressate geometricamente, possano creare problemi negli algoritmi di integrazione delle equazione del moto.
Una configurazione molecolare si trova in un minimo locale della sua superficie di energia potenziale (V) se si verifica la seguente condizione:

dove N è il numero totale di atomi del sistema, 

Tra i metodi di ricerca dei minimi locali quelli più usati sono:
- Il metodo dello steepest descent.
- Il metodo dei gradienti coniugati (conjugate gradient).
Nel primo caso, data una geometria molecolare, definita dalle coordinate 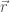


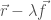



Il metodo dei gradienti coniugati parte come quello dello steepest descent, ma dopo aver individuato il minimo 

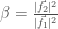



con 

