- Lex. I. Corpus omne perseverare in statu suo quiescendi vel movendi uniformiter in directum, nili quatenus a viribus impressis cogitur statum illum mutare.
- Lex. II. Muationem motus proportionalem esse vi motrici impressae, et fieri fecundum lineam rectam qua vis illa imprimitur.
- Lex. III. Actioni contrariam semper et equalem esse reactionem: sive corporum duorum actiones in se mutuo semper esse aequales et in partes contrarias dirigi.
Isaac Newton. Philosophiae Naturalis Principia Mathematica. London, 1686.
Introduzione
Nel 1648 Isaac Newton diede alle stampe la sua prima edizione dei Principia Mathematica, uno dei più grandi capolavori scientifici di tutti i tempi. Alla pagina 12 di questa summa scientifica si trovano enunciate le famose tre leggi che portano il suo nome, dalle quali ha avuto inizio la fisica-matematica classica. A 350 anni da quell’evento, le stesse leggi, usate per descrivere il moto delle stelle e dei pianeti, ci tornano ancora utili per semplificare la descrizione del mondo atomico.
Nei primi decenni dello scorso secolo, la nascita della meccanica quantistica, ha segnato l’inizio dell’esplorazione teorica della realtà a livello atomico. L’equazione di Schrödinger, al pari delle equazioni di Newton, ha permesso di riassumere in forma matematicamente elegante e coincisa, le brillanti intuizioni teoriche e i dati sperimentali accumulati nei precedenti decenni. In linea di principio, questa equazione, pur potendo essere usata per descrivere il comportamento chimico-fisico di qualunque sistema molecolare, risulta impossibile da risolvere analiticamente, quando il numero degli elettroni in gioco è superiore a due. La nascita dei calcolatori elettronici nel secondo dopo guerra, ha permesso di risolvere numericamente questa equazione per sistemi poliatomici. Tuttavia, anche con il continuo e rapido sviluppo della potenza degli elaboratori, l’equazioni di Schrödinger non può essere ancora usata per descrivere la dinamica di sistemi molcolari formati da centinaia o migliaia di atomi, quali le macromolecole biologiche. Per questo motivo, si è cercato di ridurre, attraverso opportune approssimazioni, la descrizione del comportamento dinamico degli atomi a un modello classico, in cui gli elettroni (gli elementi quantistici) non sono esplicitamente considerati ma si considera il loro effetto medio. Nasce cosi, circa 40 anni fa, la Dinamica Molecolare (DM). Grazie al continuo sviluppo di calcolatori sempre più veloci, questa tecnica si è progressivamente sviluppata estendendo il suo campo di applicazione dalla simulazione di fluidi semplici a complessi sistemi biologici, quali proteine e membrane cellulari. In tal modo, la DM sta diventando un potente e flessibile strumento d’indagine negli ambiti più disparati della biologia strutturale e della scienza dei materiali.
In questa serie di articoli verranno fornite le basi teoriche e applicative di questa tecnica.
La Dinamica Molecolare
La Dinamica Molecolare (DM) è una tecnica della chimica computazionale che consente di simulare il moto dei singoli atomi in sistemi atomici o molecolari. La descrizione completa del moto di un sistema di particelle può essere fatta usando l’equazione di Schröedinger dipendente dal tempo

dove l’operatore di Hamilton 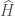




1. Breve storia della Dinamica Molecolare
Le prime simulazioni di dinamica molecolare furono effettuate negli anni 
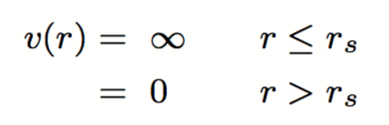
NOTA: Per dischi o sfere rigide si intendono particelle che danno luogo a collisioni perfettamente elastiche in due o tre dimensioni spaziali. Particelle di questo tipo interagiscono con un potenziale hard core, ovvero descritto dalle seguenti equazioni: con 
Il successivo uso di funzioni di potenziale più realistiche ha permesso di ottenere dati simulati direttamente confrontabili con dati sperimentali, mostrando come la DM possa essere usata come un valido strumento di indagine delle proprietà microscopiche di sistemi reali. Le prime simulazioni di questo tipo furono effettuate da Rahman e Verlet [Rahman64]: in queste simulazioni fu utilizzato un potenziale di tipo Lennard-Jones per descrivere le interazioni atomiche dell’argon allo stato liquido. Lo sviluppo di calcolatori più veloci e versatili ha consentito la simulazione di sistemi fisici più complessi. Si iniziarono, quindi, a studiare sistemi come l’acqua [Rahman71], alcani flessibili [Ryckaert75] e piccole proteine, come, per esempio, la BPTI (Bovine Pancreatic Tripsin Inhibitor) [McCammon77]. Inoltre, nello stesso periodo, furono sviluppati nuovi metodi per calcolare in modo più accurato le interazioni elettrostatiche nello studio di sistemi ionici [Woodcock71]. La simulazione di biomolecole è iniziata alla fine degli anni ’70, ed i successi ottenuti nel riprodurre dati sperimentali strutturali di proteine e macromolecole in generale, hanno portato, negli anni successivi, a una grande diffusione della DM nell’ambito degli studi di biochimica strutturale. Il continuo aumento della potenza dei calcolatori, ha consentito, in questo specifico settore, di simulare biomolecole sempre più grandi e, con l’inclusione di modelli di solvente, in condizioni sempre più realistiche [vanGunsteren90]. Attualmente la tecnica è in continuo sviluppo e la complessità dei sistemi studiati, cosi’ come la lunghezza delle simulazioni, è legata alla potenza dei calcolatori disponibili: sistemi contenenti fino a 
2. Limiti e approssimazioni
Come tutte le tecniche di calcolo con cui si studiano modelli di sistemi fisici reali anche la DM è soggetta ad assunzioni e approssimazioni che ne limitano l’accuratezza. Pertanto è necessario conoscere e valutare molto bene questi limiti per evitare di sovrastimare le capacità del metodo e quindi i risultati che questo può fornire. I problemi fondamentali che si devono considerare nelle simulazioni di DM si possono riassumere in due aspetti fondamentali:
- L’accuratezza del modello molecolare che viene usato per simulare il sistema.
I modelli fisico-matematici che descrivono la materia possono essere classificati in base al livello di dettaglio con cui descrivono il sistema che si sta studiando. Nella Tabella I sono riportati i vari livelli di approssimazione in ordine di complessità decrescente. Aumentando la complessità del fenomeno da studiare, diminuisce il livello di dettaglio con cui il sistema viene descritto attraverso l’omissione della esplicita rappresentazione di opportuni gradi di libertà. Come è stato già detto nel precedente paragrafo, quando si escludono le reazioni chimiche, le basse temperature o i dettagli del moto degli atomi di idrogeno, si può ragionevolmente assumere che il moto degli atomi del sistema possa essere descritto dalle leggi della meccanica classica (seconda riga). Nella DM classica, le interazioni tra gli atomi sono descritte da un potenziale di interazione effettiva. In questi potenziali, l’effetto medio del grado di libertà elettronico omesso è stato incorporato nel grado di libertà atomico esplicitamente presente nel modello.
- Le dimensioni dello spazio delle fasi accessibile al sistema simulato.
La simulazione di un sistema molecolare, a temperature maggiori dello zero assoluto, genera un insieme di configurazioni rappresentative di un particolare insieme statistico (vedi Capitolo VIII). Le proprietà termodinamiche di tale sistema sono definite dal loro valor medio, valutato sull’insieme delle configurazioni generate. Per l’ipotesi ergodica, la media su tali traiettorie su un tempo infinito:
dove il tempo totale durante il quale la quantità 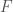

3. Struttura di un programma di Dinamica Molecolare
Nella Figure 1, è riportato in modo schematico la struttura di un programma di DM. Dopo la lettura delle coordinate iniziali e la definizione dei parametri per la simulazione, si procede dapprima alla generazione delle velocità iniziali (ottenute da una distribuzione di Maxwell-Boltzmann alla temperatura desiderata) e quindi si entra in un ciclo di calcolo la cui lunghezza dipende dal tempo di simulazione che si vuole ottenere. In questo ciclo vengono prima calcolati i potenziali e quindi, dalle derivate di quest’ultimi, le forze che agiscono su ciascun atomo. Le forze vengono quindi integrate per ottenere le posizioni e le velocità al tempo successivo. Coordinate, velocità, forze e potenziali vengono memorizzate su disco con una certa frequenza che dipende dalla accuratezza con cui si vogliono ottenere informazioni sulla traiettoria. Nei capitoli successivi verranno approfonditi gli aspetti teorici e pratici dei vari blocchi di questo schema.



Pingback: Note di Meccanica Classica | Danilo Roccatano