Italia anno 1989, era l’era del Commodore Amiga con la sua demo di BOING, la pallina bianca e rossa che rimbalzando ha conquistato il cuore di milioni di giovani che hanno vissuto la rivoluzionaria invenzione del personal computer.

Fonte: http://www.geekometry.com/2014/07/blast-from-the-past-the-amiga-boing-ball-demo-animated-gif/
Era il tempo in cui un computer crash era poeticamente chiamato GURU Meditation, dove un disco rigido di 60 MB era considerato una lussuria di abbondanza e un invidiabile privilegio.

L’epoca della programmazione in Amiga BASIC. Ho sempre un ricordo nostalgico di quegli anni. Ho passato tantisime ore nello sviluppare programmi in questo semplice e efficace ma oramai dimenticato linguaggio. Qualche anno fa, in un momento di nostalgia del passato, ho speso un po’ di tempo in uno sforzo di archeologia forense nel recuperare i prodotti perduti della mie prime esperienze di programmazione dalla mia collezione, vecchia di 20 anni, di Amiga floppy disks. Ho venduto il mio glorioso e potente Amiga 500 (era equipaggiato con una CPU 68020 con coprocessore aritmetico e un HD esterno) per sostituirlo con un PC dotato del sistema operativo Linux che conquisto’ rapidamente il mio interesse nel momento in cui cominciai ad usarlo quando iniziai il mio dottorato di ricerca nel 1993. Non più possedendo il mio Amiga 500, non potevo leggere i dischi Amiga da 800 kB con un normale PC floppy driver. Infatti, questi dischi richiedono uno speciale floppy driver poiché utilizzano un sistema di memorizzazione dei dati diverso rispetto ai normali floppy disk per PC. Avevo quasi deciso di acquistare un Amiga 500 di seconda mano su eBay (un’idea che mi passa di tanto in tanto per la mente) quando scoprii il programma Adfread creato da Toni Wilen e Simon Owen che, ingegnosamente usando due floppy driver per PC, riesce a per leggere dischi in formato Amiga. Ho avuto la fortuna di avere tra le mani un vecchio PC con una scheda madre che supporta due floppy drivers. Il programma ha funzionato benissimo con i miei dischi vecchi di 20 anni e, cosi’, ho potuto recuperare tutto il loro contenuto e in particulare i programmi che sviluppai per Amiga. Il passo successivo e’ stato quello di usare l’emulatore Amiga FS-UEA (https://fs-uae.net) con cui ho potuto emulare molto bene un Amiga sul mio laptop MacBook. L’emulazione mi ha permesso di girare i miei programmi su un Amiga 4000 (68040 CPU), il livello più alto della famiglia Amiga, nel lontano 1989 per me era un un computer da sogno dato che non potevo permettermi di acquistarlo. Questo mi ha permesso di eseguire il software che ho sviluppato più di 27 anni fa! In questo blog, descrivero’ un programma che ho sviluppato sull’Amiga e di cui sono ancora molto orgoglioso.
LA VISUALIZZAZIONE MOLECOLARE
La struttura tridimensionale di composti organici e inorganici, qualora disponibili in forma cristallina, viene determinata mediante le tecniche di diffrazione dei raggi X. L’elaborazione dei diffrattogrammi ottenuti da questi misurazioni permette di ricavare le coordinate degli atomi presenti nella unita’ fondamentale del reticolo cristallino: la cella elementare. Tali coordinate sono quelle dell’unita’ asimmetrica (cioe’ la piu’ piccola parte della molecola da cui, tramite operazioni di simmetria, e’ possibile generare la structtura completa del cristallo). Queste coordinate sono relative a un sistema di riferimento solidale con i vettori unitari che definiscono la cella elementare e sono misurate rispetto alle unita’ metriche di tali assi (da qui il nome di coordinate frazionarie). E’ raro che tale sistema di riferimento sia al tempo stesso ortogonale e monometrico (ovvero corrispondente a una cella cubica) per cui per rappresentarle e’ necessario dapprima convertirle in coordinate ortonormali con una opportuna matrice di trasformazione. Se la molecola possiede degli elementi di simmetria interni allora l’unita’ asimmetrica potrebbe representare solo una parte della struttura molecolare. In questo caso, si rende necessario, per rappresentare l’intera molecola, generare le parti simmetriche mediante opportune trasformazioni di simmetria. Un’altra importante informazione e’ il modo in cui piu’ molecole sono disposte nel cristallo, ovvero l’impaccamento cristallino. Anche in questo caso occorre dapprima generare tutti gli atomi presenti della cella elementare, e quindi applicare l’operazione di translazione della cella elementare lungo i tre assi.
IL PROGRAMMA
Il programma consiste di parti sviluppate interamente in linguaggio Amiga BASIC. Inizialmente le due parti furono implementate come due programmi separati e consistevano in un editore (Atomic Coordinates Editor o Co.Ed.) e un visualizzatore di strutture molecolari (Molecular Designer). Nell’ultima versione che completai nel 1991, decisi di unire i due programmi in uno che chiamai Molecular Model Builder (o MMB). Le operazioni fondamentali descritte nella introduzione vengono svolte dal ACEditor che si occupa di creare automaticamente la matrice delle connectivita’ molecolare (legami) tra i singoli atomi e la gestione delle molecole sul disco. l resto del programma svolge la principale funzione di rappresentare e manipolare le molecole preparate con l’ACEditor. Riporto di seguito un breve manuale che iniziai a scrivere per la versione finale (1.0) del programma. Le schermate del programma sono invece state realizzate recentemente usando l’emulatore Amiga FS-UAE (https://fs-uae.net).
MOLECULAR MODEL BUILDER (MMB versione 1)
All’avvio del programma sono selezionabili solo due item LOAD (dal menu Project) e INSERT (dal menu ATOMI). Se si vuole editare le coordinate di una nuova molecola si deve selezionare L, ultime item mentre se si vuole richiamare i dati di una molecola gia’ editata si usa il primo. Selezionando INSERT compare una finestra in alto in cui vengono richiesti nell’ ordine i parametri della cella (parametri metrici in Amstrong e angolari in gradi sessagesimali), il nome della molecola. Successivamente viene richiesto per ogni atomo il tipo inserendo il simbolo chimico dell’atomo (se si devono inserire atomi dello stesso tipo in sequenza e’ sufficiente, una volta inserito il simbolo del primo atomo, premere RETURN alle richieste successive) e le sue coordinate frazionarie. Gli atomi inseriti vengono numerati progressivamente. Inserite le coordinate e’ possibile modificarle e/o correggerle usando la opzione MOD/CANC dello stesso menu oppure agiungere altri atomi con AGGIUNGI o modificare i parametri della cella con CELLA. Si possono inoltre inserire delle note di descrizione tramite NOTE.

La schermata precedente mostra la finestra per selezionare il file con le coordinate molecolari. Nella finestra viene mostrato l’elenco di molecole presenti nella directory MOLECULES. Nel corso dello sviluppo del programma, ho copiato le coordinate cristallografiche frazionarie di decine di molecole da libri e riviste di cristallografia depositati dalla biblioteca del Dipartimento di Chimica dell’Universita’ la Sapienza di Roma. All’epoca ero uno studente universitario in Chimica e non potevo accedere al famoso database cristallografico Cambridge Structural Database.
Per capire come usare queste coordinate, passai del tempo studiando cristallografia (tempo ben speso poichè di li’ a poco mi fu molto utile per il corso di cristallografia!). Riuscii, e non senza fatica, a trovare la formula generale per trasformare le coordinate atomiche frazionarie a ortonormali. Ricordo che fui giubilante e orgoglioso di me stesso quando finalmente trovai la corretta forma della matrice di transformazione!
Le coordinate atomiche sono riportate come coordinate frazionarie (cioè divise per la lunghezza dei lati (a, b, c) della cella unitaria) nel riquadro di riferimento della cella unitaria. Per rappresentarli nella struttura cartesiana del sistema di riferimento utilizzato dal programma, è necessario applicare una trasformazione del sistema di riferimento che tenga conto degli angoli cristallografici (

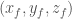
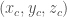

Questo e’ il pezzo di codice usato nel mio programma per effettuare questa conversione
' In Amiga Basic
' Convert Fractional to Cartesian Coordinates
X=XF*AA
Y=YF*BB
Z=ZF*CC
XC=X*SIN(ga)+Z*((COS(be)-COS(ga)*COS(al))/SIN(ga))
YC=X*COS(ga)+Y+Z*COS(al)
ZC=(Z/SIN(ga))*SQR(1-(COS(ga)^2)-(COS(al)^2)-(COS(be)^2)+2*COS(al)*COS(be)*COS(ga))
Il passo successivo consiste nel generare la matrice delle connettivita’ molecolari (o dei legami) tra gli atomi (e’ consigliabile, prima di far cio’, memorizzare la lista delle coordinate inserite con SAVE del menu PROJECT). Questa operazione puo’ essere condotta automaticamente con l’opzione AUTOGENERA del menu LEGAMI oppure manualmente con l’opzione COR.LEG. dello stesso menu (questa opzione va usata anche per ritoccare la matrice autogenerata). I legami vengono memorizzati nella matrice tridimensionale co%() in modo che ogni riga corrisponde ad un atomo della molecola, nella colonna di indice 0 viene memorizzato il numero di legami dell’atomo, e nelle colonne succesive i numeri degli atomi a cui si lega, nel secondo livello della matrice in corrispondenza di ogni legame viene inserito il tipo di legame (1=semplice, 2=doppio, 3=triplo, tali valori vanno inseriti alla richiesta del tipo di legame quando con l’opzione COR.LEG. si modifica la marice co%).
Non appena si genera la matrice co% viene mostrata sullo schermo la rappresentazione a linee (wireframe) della molecola.
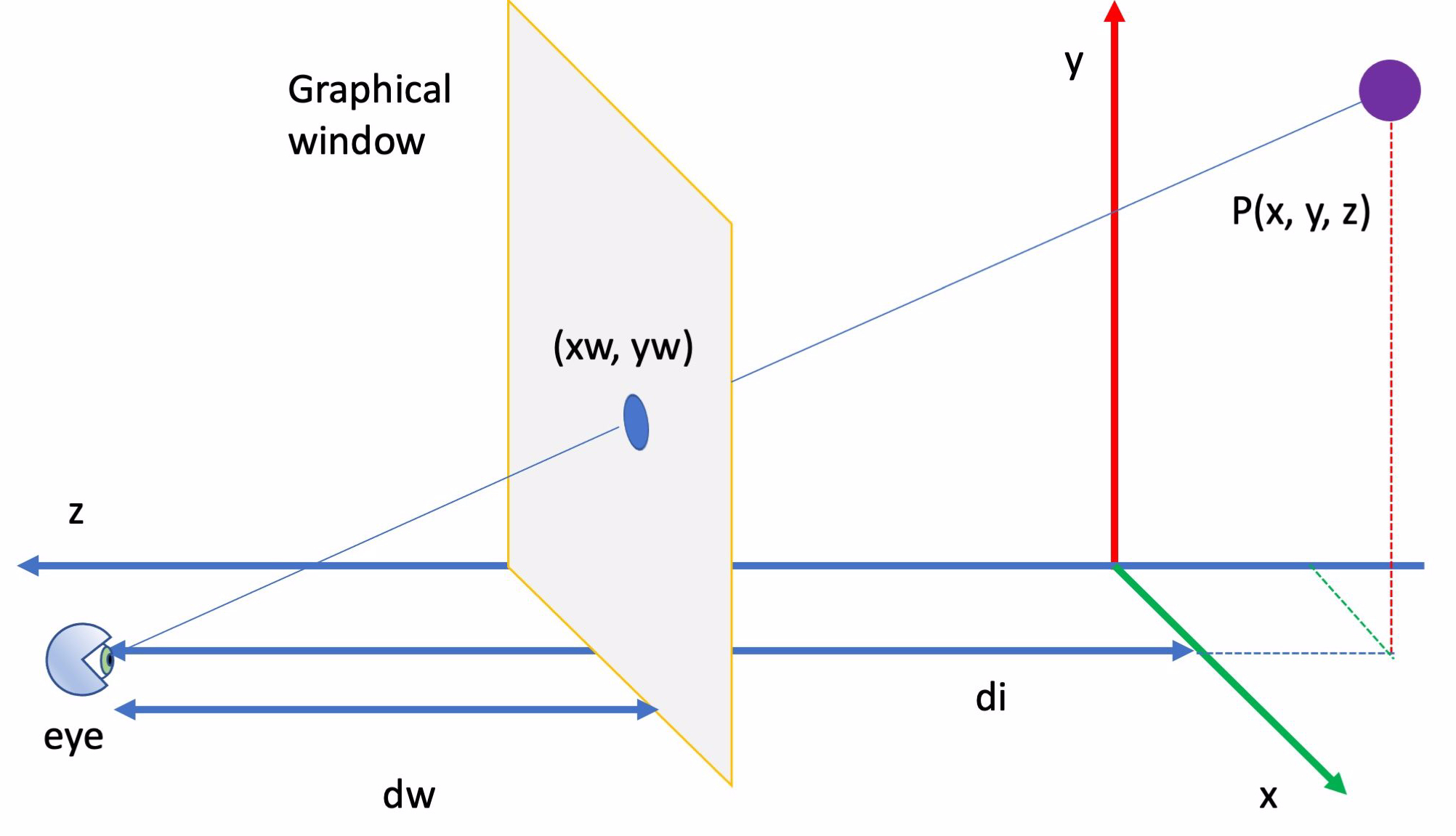
Le coordinate tridimensionali della molecola vengono proiettate nella finestra di visualizzazione usando le seguenti formule di proiezione


con 
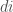
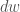

La cella unitaria può anche essere visualizzata con le coordinate atomiche. Nello screenshot qui sopra, vengono mostrate la struttura cristallina della molecola di beta-carotene e la cella unitaria cristallografica.
A questo punto si puo’ agire sul menu OP.GRAFICHE per la manipolazione dell’immagine. Prima di parlare dei comandi grafici finisco di descrivere gli altri menu. Il menu OUTPUT contiene una serie di item che gestiscono la rappresentazione dei dati elaborati dal programma. Esso e’ diviso in tre parti. La prima parte serve a selezionare la device su cui dirigere l’output (schermo, printer, disco). La seconda serve a selezionare le informazioni da mandare in output.Infine l’ultima parte contiene il comando per l’esecuzione dell’output. L’item GEN.SIM. serve per impostare delle matrici che effettuano le trasformazioni isometriche delle coordinate inserite, permettendo, in tal modo, di generare parti simmetriche della molecola. L’opzione Packing genera l’impaccamento della molecola nel cristallo. Selezionando tale comando viene visualizzato il numero di celle generabili e poi viene richiesto il numero di celle per asse (oltre quella di base).
Nel menu PROJECT e’ presente l’opzione LOAD che consente di richiamare i dati di una molecola memorizzata sul disco. Selezionando LOAD compare una finestra con un riquadro a scorrimento in cui sono visualizzati i nomi delle molecole presenti nella directory MOLECULES. I dati relativi ad una molecola vengono memorizzati nella stessa directory (che deve essere sempre presente sul disco di lavoro) in un file sequenziale denominato con un numero progressivo di 5 cifre max. Questo numero unito al nome della molecola viene inserito nel file indice LISTA che viene letto al momento dell’avviamento del programma (puo’ essere riletto, se si cambia il disco con i dati, usando l’item L.LISTA) e visualizzato (solo i nomi delle molecole) nel riquadro del file request. Se i nomi visualizzati eccedono in lunghezza appare lateralmente ad essi due freccette di scorrimento orizzontale che consentono la visione completa. Per selezionare una molecola e’ sufficiente cliccare con il mouse sul nome. In basso sono presenti 5 tasti
- RENAME: serve a cambiare il nome di una molecola.
- PRINT: stampa il contenuto del file lista
- LOAD: carica i dati della molecola selezionata.
- CANCEL: esce dal filerequest.
che svolgono le funzioni indicate sul file selezionato.
OPERAZIONI GRAFICHE
Nel menu PROJECT, il commando DUMP effettua la stampa della schermata. Nel menu OP.GRAFICHE sono presenti i commandi di rotazione (o RUOTA X,Y,Z) che consentono la rotazione della molecola intorno agli assi di un certo angolo a partire da un angolo iniziale (questi parametri possono essere impostati con l’opzione PARAMETRI). Le matrici di rotazione elementari per la rotazione lungo l’asse x, y e z sono




Le due opzioni ZOOM IN, OUT permettono di ingrandire o rimpicciolire la molecola , anche in questo caso lo step di zoom viene fissato con PARAMETRI. E’ possibile autodimensionare la figura agendo sull’opzione AUTODIMENS., inoltre e’ possibile la centratura automatica con CENTRA. L’item TRASLAZ. permette di spostare con il mouse la proiezione del baricentro della molecola (la crocetta) sullo schermo, spostando di conseguenza l’intera molecola.
MENU RAPP
In questo menu sono presenti una serie di comandi che consentono di variare il modo di rappresentare graficamente le molecole. Il modo di default di rappresentazione delle molecole e’ wireframe (cioe’ vengono visualizzati i legami con delle linee) a colori.
- NOMI ATOMI: visualizza i simboli chimici degli atomi che compongono la molecola.
- ATOMO NUMERATI: visualizza il numero d’ordine di ciascun atomo.
- ATOMI B/N: traccia la struttura wireframe in bianco e nero.
- ATOMI COLOR: traccia la struttura wireframe a colori.
- SPACE-FILLING: per ogni atomo disegna un cerchio di raggio proporzionale al raggio di Van der Walls in tal modo si riesce ad avere un’idea del volume reale occupato dalla molecola.
- STICK & BALLS: disegna la molecola visualizzando i legami come bastoncini
e gli atomi come sferette (ball&stick). - DIS.CELLA: disegna (con la molecola nella struttura ,e solo in questa,di tipo wireframe) la cella elementare.
- EL.IDROGENI: visualizza la molecola senza i legami con gli atomi d’idrogeno eventualmente presenti.
- EL.SCRITTE: abilita e/o disabilita le scritte sullo schermo.
- LEG.MULTIPLI1: evidenzia i legami multipli con doppia o tripla linea a seconda che siano doppi o tripli.
- LEG.MULTIPLI2: evidenzia i legami multipli con tratteggio differenziato.
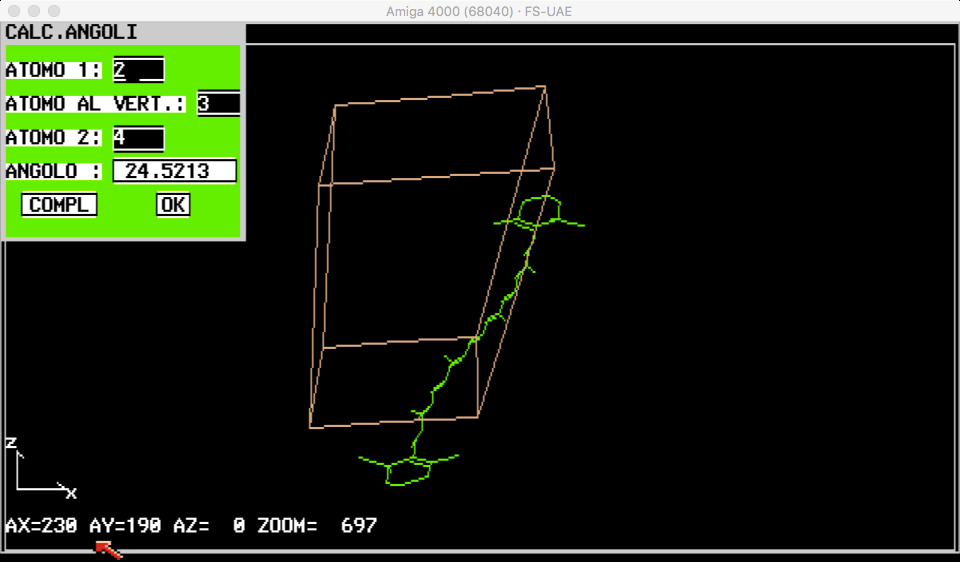
MENU EXTRAS. Questo menu permette il calcolo di distanze o angoli tra atomi.
- ANGOLI: visualizza un request in cui ,inserendo i numeri d’ordine di tre atomi negli appositi punti, si ottiene il valore dell’angolo minimo da essi formato (ed anche il valore complementare)
- DISTANZE: con questo opzione si puo’ conoscere la distanza (in amstrong) tra
due atomi qualsiasi della molecola.
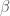 -carotene nella cella unitaria.
-carotene nella cella unitaria. MENU SCHERMO
- BIANCO: rende lo sfondo bianco.
- NERO: rende lo sfondo nero.
- LEGENDA: visualizza i colori attribuiti in modo standard a 12 tipi di atomi
(gli altri atomi vengono visualizzati con uno stesso colore):- Idrogeno ⇒ GRIGIO
- Boro ⇒ ROSSO
- Carbonio ⇒ VERDE
- Azoto ⇒ BLU SCURO
- Ossigeno ⇒ ROSSO CILIEGIA
- Fluoro ⇒ BLU CIELO
- Zolfo ⇒ GIALLO
- Fosforo ⇒ ARANCIO
- Cloro ⇒ VERDE LIMO
- Bromo ⇒ ACQUA
- Iodio ⇒ ROSSO FERRARI
- Altri tipi di atomo ⇒ MARRONE

- INTERLAC: abilita e/o disabilita schermo interlacciato.
Questo e’ il listato del programma MMB v.1.0: mmb1

