Questo blog contiene un estratto del capitolo introduttivo sulla spettroscopia XANES della mia tesi di laurea che discussi 25 anni fa, per cui gli argomenti non sono molto aggiornati anche se il contenuto riportato è di carattere generale.
Spettri di assorbimento di raggi X
Fino agli inizi del 1970 c’è stata una netta distinzione tra le tecniche di spettroscopia di assorbimento e quelle di diffrazione dei raggi X. La diffrazione era tradizionalmente considerata lo strumento più importante per la determinazione della struttura atomica di un composto, mentre l’assorbimento e l’emissione di raggi X erano usati per comprenderne la struttura elettronica. Questa distinzione fu definitivamente superata quando, nel 1971 Sayers e collaboratori (Sayers et al., 1971) mostrarono che era possibile ricavare informazioni sulla disposizione degli atomi circostanti un atomo assorbitore, analizzando la struttura fine degli spettri di assorbimento ai raggi X. La possibilità di selezionare la specie atomica eccitata, variando l’energia dei fotoni incidenti, e di analizzare gli stati elettronici finali eccitando diversi livelli di core, ha reso la tecnica spettroscopia di assorbimento dei raggi X un ottimo mezzo d’indagine per la struttura locale della materia. Fino all’inizio del 1990, i raggi X erano prodotti usando sorgenti tradizionali (tubi catodici). La successiva disponibilità di sorgenti di luce di sincrotrone (Daresbury, 1981) ha dato un sostanzialmente contribuito all’ulteriore sviluppo e diffusione di questa tecnica. Nei paragrafi che seguono, sarà data una succinta introduzione alla tecnica della spettroscopia ai raggi X.
L’ assorbimento dei raggi x nei sistemi condensati
Uno spettro di assorbimento si ottiene illuminando il campione della sostanza in esame con un fascio monocromatico di fotoni (ovvero aventi un certo valore di energia) e misurando l’attenuazione della sua intensità dopo il passaggio. L’attenuazione varia monotonamente con l’energia dei fotoni incidenti eccetto che per alcuni valori in corrispondenza dei quali si hanno dei salti d’assorbimento come quello a circa 8980 eV mostrato in Figura 1. Questi valori sono denominati soglie di assorbimento e corrispondono all’energia necessaria al fotone per eccitare gli elettroni dell’atomo assorbitore dai livelli elettronici di core a quelli esterni non occupati che si estendono tra gli stati di valenza e il continuo.
 Figura 1. Spettro di assorbimento XANES della soglia K del rame. E’ mostrata approssimativamente la divisione tra la “regione XANES” e la “regione EXAFS”. La regione di soglia comprende circa 8 eV attorno alla soglia di assorbimento (8985.4 eV).
Figura 1. Spettro di assorbimento XANES della soglia K del rame. E’ mostrata approssimativamente la divisione tra la “regione XANES” e la “regione EXAFS”. La regione di soglia comprende circa 8 eV attorno alla soglia di assorbimento (8985.4 eV).
Gli spettri della soglia K sono dovuti alle transizioni 1s → p e perciò descrivono gli stati finali con l=1 (per la regola di selezione in approssimazione di dipolo elettrico Δl= ± 1), in altre parole la densità degli stati finali di orbitali p non occupati.
Lo spettro di assorbimento fino a circa 1 keV dopo la soglia non varia monotonamente con l’energia, ma ha un comportamento che si può suddividere qualitativamente in tre parti:
- La regione di soglia, che si estende per circa 8 eV, dove si osservano strutture (picchi d’assorbimento) deboli (come nel caso della soglia K del Fe) dovute a transizioni di dipolo e quadrupolo dal livello 1s agli orbitali molecolari.
- La regione XANES (X-ray Adsorption Near-Edge Structure) si estende per circa 50 eV dopo la regione di soglia ed è caratterizzata da strutture intense.
- La regione EXAFS (Extended X-ray Adsorption Fine Structure) si estende per circa 1 keV dopo la regione XANES ed è caratterizzata da oscillazioni multiple.
Questo tipo di comportamento appare solo quando gli atomi sono nello stato condensato. Per atomi isolati, l’assorbimento dopo la soglia varia in modo monotono. Le strutture osservate in fase condensata sono dovute all’interazione del fotoelettrone con gli atomi che circondano quello emettitore. Come discuteremo è possibile, elaborando questi dati, ottenere informazioni sulla struttura locale intorno all’atomo foto assorbitore.
Coefficiente di assorbimento
L’attenuazione dei raggi X dovuta al passaggio in un materiale segue la legge di Beer-Lambert:

dove 
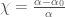
dove 


dove n è il numero di atomi assorbitori per unità di volume, pertanto esso permette di dedurre informazioni sulla disposizione degli atomi nel campione. L’attenuazione della radiazione elettromagnetica al passaggio in un materiale è causata principalmente a tre fenomeni: diffusione, assorbimento fotoelettrico e produzione di coppie, le cui sezioni d’urto variano diversamente in funzione dell’energia.
Nella regione dei raggi X (
La seguente descrizione teorica del fenomeno XANES è basata sull’ approssimazione di singolo elettrone – ovvero considerando la foto eccitazione di un solo elettrone che lascia una buca elettronica nello stato di core. Per il momento, le eccitazioni elettroniche multiple sono trascurate. In tal modo, il processo può essere descritto in approssimazione semi-classica, considerando il fotone come un campo elettromagnetico classico e l’elettrone come una particella quantistica. Se la lunghezza d’onda del fotone X incidente è maggiore rispetto alle dimensioni dello stato elettronico di core eccitato, il campo elettromagnetico può essere considerato costante nella sua sovrapposizione con lo stato di core e può essere approssimato da un potenziale del tipo 

dove $latex E_i$ ed $latex E_f$ sono le energie dello stato iniziale e finale, a è la costante di struttura fine, E è l’energia del fotone incidente, p è la sua polarizzazione, D è l’operatore di dipolo elettrico e la funzione delta di Dirac 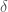
Pertanto, la probabilità che un fotone sia assorbito da un elettrone di core dipende dallo stato iniziale e da quello finale dell’elettrone. Lo stato iniziale è fissato al livello di core localizzato in corrispondenza alla soglia di assorbimento. Pertanto, il contributo oscillante al coefficiente di assorbimento può dipendere solo dallo stato finale del processo, cioè dallo stato del fotoelettrone. Nei paragrafi che seguono verrà spiegato il modo in cui questo avviene.
Regione EXAFS
Nei sistemi condensati gli stati elettronici più facilmente descrivibili sono quelli di bassa e di alta energia. A bassa energia abbiamo gli stati di core, che sono essenzialmente uguali a quelli degli atomi del sistema, ad alta energia abbiamo gli stati del continuo, dove gli elettroni sono approssimabili con onde piane debolmente diffuse dagli atomi del sistema. Questa è la regione che da luogo alla parte EXAFS nello spettro di assorbimento dei raggi X.
Per semplicità assumiamo inizialmente che il fotoelettrone abbia un’energia cinetica grande rispetto all’energia d’interazione con altri atomi e cioè poniamoci a circa 50 eV sopra la soglia. In questo modo lo si può descrivere come un’onda sferica uscente dall’atomo assorbitore e trattare l’interazione con gli atomi circostanti come una perturbazione. Pertanto, il fotoelettrone uscente sarà diffuso indietro dagli atomi vicini producendo da ognuno di essi un’onda elettronica sferica, com’è illustrato in Figura 2. Lo stato finale sarà quindi la somma risultante dell’onda elettronica uscente e di tutte quelle rientranti l’atomo foto-assorbitore. L’interferenza di queste onde è quindi l’origine delle variazioni periodiche del coefficiente di assorbimento. Infatti, il cambiamento della funzione d’onda del fotoelettrone modifica la sua sovrapposizione con lo stato di core nell’elemento di matrice di dipolo dell’equazione (4) e quindi il valore del coefficiente d’assorbimento. Inoltre, la differenza di fase tra l’onda entrante e quella uscente varia al variare dell’energia del fotone X e quindi l’interferenza varia da costruttiva a distruttiva al variare dell’energia.
Il coefficiente d’assorbimento quindi quantifica l’interferenza dello stato finale del fotoelettrone con se stesso. Il modo in cui varia la fase di queste onde dipende dalla distanza tra l’atomo assorbitore e quelli diffusori mentre l’intensità dell’onda diffusa dipende dal tipo di atomo diffusore. Pertanto, il segnale EXAFS contiene informazioni sulla geometria e natura chimica degli atomi attorno all’atomo centrale.
Regione XANES
Nella materia condensata gli stati che legano il sistema e che ne determinano le proprietà elettroniche sono quelli che si estendono a energie comprese tra lo stato di core e quello non legato e cioè tutti i livelli non occupati dal quello di Fermi fino a quelli prossimi al continuum. Questi vengono anche detti stati XANES poiché sono i livelli che sono popolati dagli elettroni eccitati con fotoni di raggi X il cui valore di energia si estende dal valore di soglia fino a circa 50 eV al sopra di essa.

Figura 2. Rappresentazione schematica dell’interferenza tra la porzione entrante e quella uscente della funzione d’onda del fotoelettrone emesso dall’atomo assorbitore.
Nella regione EXAFS, l’interazione tra gli elettroni eccitati e gli atomi è così debole che l’unico contributo rilevante alla funzione d’onda dello stato finale in prossimità dell’atomo assorbitore, è dato dai cammini in cui l’elettrone ha subito una sola diffusione. Quando l’energia del fotoelettrone è localizzata nella regione XANES, l’interazione tra l’elettrone e gli atomi aumenta e il fenomeno della diffusione multipla diventa un contributo molto importante. In termini matematici è possibile espandere la sezione d’urto (Eq. 4) come somma di contributi parziali
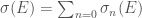
Il termine n=0 rappresenta la variazione monotona della sezione di assorbimento atomico, il termine n=1 è sempre uguale a zero, e il termine generico n è il contributo alla sezione d’urto di foto-assorbimento dal processo in cui il fotoelettrone è stato diffuso n-1 volte dagli atomi circostanti prima di ritornare sul sito foto assorbitore. Il coefficiente di assorbimento totale può essere scritto come:
![\alpha(E) = \alpha_0 \left[1+\sum_{n=2} \chi_n(E)\right]](https://s0.wp.com/latex.php?latex=%5Calpha%28E%29+%3D+%5Calpha_0+%5Cleft%5B1%2B%5Csum_%7Bn%3D2%7D+%5Cchi_n%28E%29%5Cright%5D&bg=ffffff&fg=444444&s=0&c=20201002)
dove 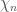
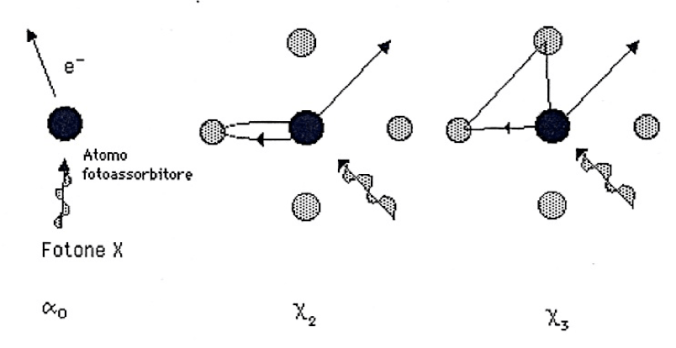
Figure 3. Rappresentazione geometrica dei primi due termini dello sviluppo in serie della probabilità di transizione $latex \sigma(E)$.
Affinché lo sviluppo di 

L’ampiezza del segnale osservato è ridotta dalla diffusione anelastica del fotoelettrone. Questo fenomeno è causato da due tipi di processi. Il primo tipo è dovuto all’eccitazioni elettroniche multiple dell’atomo centrale (multibody transitions). Infatti, l’interazione dei raggi X con gli elettroni di core dell’atomo foto assorbitore crea una lacuna elettronica, di conseguenza i N-1 elettroni non eccitati vedono un potenziale nucleare differente in quanto lo schermaggio elettronico è diminuito. Pertanto, per bilanciare la buca creata dall’eccitazione, gli elettroni dei livelli più energetici rilassano verso in quello vuoto a energia minore creando transizioni multiple dovute alla cascata di processi di rilassamento elettronico. Pertanto lo stato finale del fotoelettrone viene a essere influenzato dalla presenza di più configurazioni finali dell’atomo emettitore.
Per semplificare la trattazione del problema si ricorre all’approssimazione di singolo elettrone. In tale trattazione si assume che il fotoelettrone veda una sola configurazione statica di elettroni passivi (the fully-relaxed) definita, tra le possibili, come quella avente l’energia più bassa (von Barth e Grossman, 1982). In tal modo, per le transizioni da un livello elettronico di tipo 1s, lo stato iniziale e finale possono essere fattorizzati come segue:

dove 
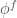


dove il primo termine da luogo ad un integrale di sovrapposizione che è minore di uno e quindi semplicemente riduce l’ampiezza dell’assorbimento del singolo elettrone (Teo, 1981; Bianconi et al., 1991).
Il secondo tipo processo è la diffusione anelastica del fotoelettrone rispetto agli elettroni di valenza. Questo secondo effetto porta a un tempo di vita media 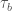
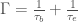

Nel regime EXAFS dello spettro, dominato dal single scattering, l’informazione strutturale è facilmente estraibile come funzione di distribuzione radiale effettuando la trasformata di Fourier delle oscillazioni del coefficiente di assorbimento (Stern e Zherig, 1988). Nel regime XANES, il segnale è dominato dal fenomeno del multiple scattering fotoelettronico che conferisce al segnale una sensibilità strutturale non soltanto alle distanze radiali ma anche agli angoli di legame (Durham, 1988). Inoltre, nella regione di soglia, variazioni nella distribuzione di carica attorno all’atomo fotoemettitore, per via di cambiamenti del suo intorno chimico, possono alterare le energie di legame dei livelli di core producendo degli spostamenti energetici della soglia di assorbimento che sono rilevabili nello spettro XANES.
La tecnica XANES è stata utilizzata in numerosi studi condotti su molecole biatomiche, metalli, isolanti, materiali amorfi e cristallini (Bianconi, 1988). Il livello di raffinatezza raggiunto permette di estendere questi studi a materiali più complessi in particolare a molecole biologiche. I vantaggi di queste tecniche per questo tipo di studi sono i seguenti:
- A differenza della tecnica di diffrazione dei raggi X, non è necessario che la struttura abbia un ordine a lungo raggio e quindi può essere usata anche per studiare sistemi in soluzione oltre che a quelli allo stato solido.
- Le strutture nella regione XANES dello spettro sono normalmente molto più intense delle oscillazioni nella regione EXAFS e quindi permettono di studiare la struttura in sistemi in soluzione con maggiore dettaglio rispetto alla seconda.
- Le stesse strutture locali in sistemi con diverso ordine a lungo raggio possono avere uno spettro XANES molto simile ed è quindi possibile usare molecole modello più semplici per studiare la struttura locale presente in molecole più complesse (ad esempio l’intorno dell’heme nelle proteine) (Bianconi, 1988). In questo studio di tesi abbiamo considerato un composto che può essere usato come modello della struttura del sito attivo di varie proteine contenenti rame (ad esempio la proteina superossido dismutase).
- La distribuzione atomica locale può essere determinata separatamente attorno a ciascun tipo di atomo ottenendo informazioni sia riguardo alle distanze, che agli angoli di legame.
- La misura XANES effettuata con il metodo dispersivo, è molto rapida con tempi che possono avvicinarsi a quelli delle reazioni biologiche. In questo modo è possibile studiare i meccanismi di queste reazioni.
Come si è già detto nell’introduzione, il maggiore problema della spettroscopia a raggi X risiede nell’analisi dei dati ovvero nella estrazione della pletora di dati strutturali ed elettronici contenuti nei dati sperimentali. La procedura per ottenere le caratteristiche strutturali dai dati sperimentali prevede il confronto dello spettro misurato con quello teorico calcolato da un modello strutturale del sistema. Essendo sia lo XANES sia l’EXAFS, tecniche per studiare strutture molecolari locali non possono sostituirsi alle tecniche di diffrazione, ma possono esserne dei validi complementi. In particolare, lo spettro XANES, essendo la più sensibile dei due, può fornire risultati molto accurati di distanze e angoli di legame ma è anche molto sensibile alle piccole variazioni di questi valori. Esso può essere usata sia nella scelta tra i diversi modelli proposti in base agli altri esperimenti, sia per determinare i cambiamenti indotti nelle strutture rispetto ai composti modello (Bianconi, 1988). Questo rappresenta un importante vantaggio della tecnica ma anche una delle maggiori difficoltà nell’elaborazione dei dati sperimentali poiché il modello molecolare usato per il calcolo dello spettro teorico deve essere anche molto accurato. Normalmente il modello iniziale è derivato dalla struttura cristallografica della molecola in esame o di composti di simile struttura. Dal modello si calcola quindi lo spettro teorico che è confrontato con quello sperimentale. Se necessario, l’accordo è migliorato con successive variazioni nella posizione degli atomi intorno a quello foto-assorbitore. Purtroppo, questa procedura, oltre ad essere tediosa se effettuata manualmente, non tiene conto dell’effetto della mobilità atomica e pertanto non fornisce una descrizione accurata dei contributi dinamici allo spettro sperimentale.


Pingback: Il Trigesimo Anniversario della Tesi di Laurea |