Write in C (Let it Be)
When I find my code in tons of trouble,
Friends and colleagues come to me,
Speaking words of wisdom:
“Write in C.”
As the deadline fast approaches,
And bugs are all that I can see,
Somewhere, someone whispers:
“Write in C.”
Write in C, write in C,
Write in C, oh, write in C.
LISP is dead and buried,
Write in C.
I used to write a lot of FORTRAN,
For science, it worked flawlessly.
Try using it for graphics!
Write in C.
If you’ve just spent nearly 30 hours
Debugging some assembly,
Soon you will be glad to
Write in C.
Write in C, write in C,
Write in C, yeah, write in C.
Only wimps use BASIC.
Write in C.
Write in C, write in C,
Write in C, oh, write in C.
Pascal won’t quite cut it.
Write in C.
Write in C, write in C,
Write in C, yeah, write in C.
Don’t even mention COBOL.
Write in C.
Parody Song by
Brian Marshall
This series of tutorials will provide a short and practical introduction to the C language aiming students with interest in computational or physical chemistry. At the end of this tutorial, you will be able to write simple programs that can read data from files elaborate them and write the results of the calculations in output files. This tutorial is not a course in C programming language, therefore the motivated readers are encouraged to look for more comprehensive introductions to this language.
The tutorial is also based on OS based Unix systems such as Linux or MacOSX. Therefore, I recommed to give a look to my introductions to Unix OS:
Continue reading


 . Questi potenziali di energia intermolecolari possono essere classificati in base al tipo di interazioni che descrivono.
. Questi potenziali di energia intermolecolari possono essere classificati in base al tipo di interazioni che descrivono. 


 e
e  (con
(con  il valore dell’unita’ di carica elettronica e
il valore dell’unita’ di carica elettronica e  la carica netta dello ione) separati dalla distanza
la carica netta dello ione) separati dalla distanza 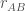 .
.



