Le forze interatomiche o intermolecolari che tengono uniti i cristalli molecolari sono di origine elettrostatica e possono essere classificate in forze a corto e lungo raggio a seconda di quanto si estende nello spazio la loro azione. Essendo funzione della distanza tra gli atomi (o molecule), queste forze sono dei campi vettoriali conservativi e sono pertanto derivabili dal gradiente di un potenziale di energia scalare, ovvero 
Interazioni tra ioni e dipoli permanenti
Interazione ione-ione
In questo caso abbiamo un potenziale di tipo Columbiano dato dalla espressione:

dove 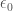



Interazione ione-dipolo
Nel caso di uno ione di carica q che interagisce con un dipolo permanente di momento 

dove 

dove 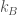
Interazione dipolo-dipolo
L’energia di interazione tra due dipoli permanenti è descritto dalla relazione:

dove gli angoli 
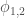

Interazioni tra dipoli indotti
In presenza di un campo elettrico esterno 

Interazioni ione-dipolo indotto
In questo caso si mostra che il valore del potenziale è dato dalla espressione:

Interazioni dipolo – dipolo indotto
In questo caso si mostra che il valore del potenziale è dato dalla espressione:

Interazioni tra due dipoli indotti
Questo tipo di interazione è presente in tutte le molecole ed è dovuto alla fluttuazione della densità elettronica, che in ogni momento crea un dipolo istantaneo il cui campo elettrico esterno induce a sua volta un momento di dipolo in una molecola vicina. Queste interazione hanno origine puramente quantomeccanica e furono per la prima volta trattate da F. London nel 1930, da cui il nome di forze di London. La frequenza di vibbrazione degli elettroni è molto alta e questo comporta che la polarizzabilità non è più una quantità statica ma è legata al moto degli elettroni sotto l’influenza di un campo elettromagnetico nella regione ottica dello spettro. Per questo motivo, queste forze sono state chiamate forze di dispersione poiché sono legate alla interazione tra la radiazione e la materia e forniscono la base per la teoria della dispersione della luce da parte dei mezzi materiali. Per due oscillatori armonici si ottiene l’espressione:

dove 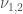


dove 
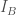
Table 1: Valore di n per diverse configurazioni elettroniche.
| Molecola | 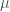 (a) (a) |
 (b) (b) |
Energia h |
Orientazione | Induzione | Dispersione |
| He | 0 | 0.20 | 24.5 | 0 | 0 | 1.2 |
| Ar | 0 | 1.63 | 15.4 | 0 | 0 | 52 |
| Xe | 0 | 4.00 | 11.5 | 0 | 0 | 217 |
| CO | 0.12 | 1.99 | 14.3 | 0.0034 | 0.057 | 67.5 |
| HCl | 1.03 | 2.63 | 13.7 | 18.6 | 5.4 | 105 |
| HI | 0.38 | 5.4 | 12 | 0.35 | 1.68 | 382 |
NH |
1.5 | 2.21 | 16 | 84 | 10 | 93 |
H O O |
1.84 | 1.48 | 18 | 190 | 10 | 47 |
(a) Momento di dipolo, in Debye (D).
(b) Polarizzabilità (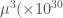
Interazione repulsiva
Quando due atomi vengono portati ad una distanza inferiore a quella della somma dei rispettivi raggi di van der Waals si generano interazioni repulsive dovute alla compenetrazione delle nuvole elettroniche dei due atomi. Le energie di interazione repulsiva vengono descritte come funzioni 
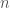

Legami idrogeno intermolecolari
Queste interazioni sono possibili quando vi sono coppie di atomi fortemente elettronegativi (N,O, alogeni) di cui uno è legato a un idrogeno. L’energia di legame è dell’ordine di 4-7 kcal/mole ed è una interazione fortemente orientante.
IL POTENZIALE DI LENNARD-JONES
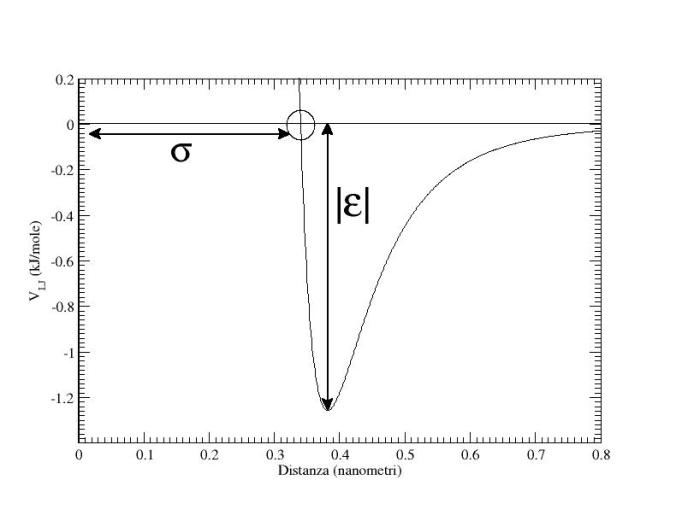
Una delle più utilizzate espressioni per la descrizione del potenziale di interazione non legame di non legame è il potenziale di Lennard-Jones. Il potenziale di Lennard-Jones è formato da due termini che descrivono una interazione di tipo repulsivo e una di tipo attrattivo:

A e B sono delle costanti empiriche che possono essere ricavate da dati sperimentali o da calcoli quantomeccanici. Una forma molto usata del potenziale di Lennard-Jones, sostituisce alle due costanti A e B, due quantità 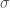

![\displaystyle V_{LJ} = 4 \epsilon \left[\left(\frac{\sigma}{r}\right)^{12} - \left(\frac{\sigma}{r}\right)^{6}\right]\hfill(12)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+V_%7BLJ%7D+%3D+4+%5Cepsilon+%5Cleft%5B%5Cleft%28%5Cfrac%7B%5Csigma%7D%7Br%7D%5Cright%29%5E%7B12%7D+-+%5Cleft%28%5Cfrac%7B%5Csigma%7D%7Br%7D%5Cright%29%5E%7B6%7D%5Cright%5D%5Chfill%2812%29&bg=ffffff&fg=444444&s=0&c=20201002)
I valori di


ANALISI CONFORMAZIONALE
Le forze intermolecolari possono agire anche intramolecolarmente contribuendo in modo sostanziale alla cosiddetta energia conformazionale. Nella stereochimica esistono tre importanti concetti: Costituzione, Configurazione e Conformazione che definiscono la struttura di una molecola. Per costituzione si indica la composizione atomica e la connessione che questi hanno, ovvero la topologia molecolare che può essere descritta mediante un grafo chimico. La configurazione indica i diversi arrangiamenti che gli atomi di una molecola di data costituzione possono avere, non considerando quelli dovuti alla rotazione intorno a legami semplici. Infine, con conformazione si fa riferimento ai diversi arrangiamenti spaziali di una molecola di configurazione data per rotazione intorno ad un legame singolo.
La rotazione dei gruppi chimici intorno ad un legame singolo determina, in conseguenza delle interazioni repulsive con gli altri gruppi della molecola, un diverso valore di energia conformazionale.


Hi nice reading your postt
LikeLike