Understanding the mechanisms of the molecular recognition has fundamental impacts in medicine and biotechnology. It plays an important role in discovering new drugs and in developing new biocatalyst. The theoretical study of these mechanisms has boosted the development of approximated but fast methods for screening large compound libraries and protein-protein complexes. Nowadays, the study of the docking of two molecular systems can be performed in full detail using very long MD simulations, this it has been possible thank to the developing of efficient programs and dedicated computers. MD simulations have the advantage to study the process by including full flexibility of the substrate and protein and using explicit solvent environment. However, the demanding calculation time still requires, for being competitive with the traditional approaches, ingenious methods to accelerate the searching of docking configurations within the timescale affordable by conventional MD simulations. An approach consists in the use of Steered Molecular Dynamics (see another article on this method in this blog) by applying a pulling force on the ligand to drive it into the active site [65]. However, this method requires the pre-knowledge of the docking site(s). When this prerequisite is missing, a simple and effective method to accelerate the docking process is to artificially increase the diffusion of the ligand around the receptor. This approach, called MDD (Molecular Dynamics Docking) method, was proposed for the first time in 1994 and it was the first example in literature of successful MD docking study in the explicit solvent [1, 2]. The MDD algorithm is based on the use of the Molecular Dynamics simulation method to increase the efficiency of the searching for protein binding sites by a substrate.
It is based on the separation of the velocity of the center of mass (c.o.m.) of the ligand from those of its internal degrees of freedom; the two velocities are coupled to two external baths, the one, coupled with the c.o.m. motion, is kept to very high temperature, whereas the one coupled to the internal motions is kept at a room temperature. In this way, the system can rapidly diffuse within the simulation box and explore more efficiently the surface of the protein.
The kinetic energy of the substrate molecule is given by

where 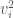

where 
The total linear momentum with respect the c.om. of the system is zero.

The kinetic energy in the reference system can be expressed as

with 

For isolated molecules the two energies are uncoupled since the c.o.m. energy can be changed only by external forces. As the substrate interacts with the receptor, the forces on the substrate by the receptor influence both the internal and the c.o.m. energies, coupling them and determining an exchange of energy between these two terms.
In this way, the substrate dissipates the kinetic energy of the c.om. along many degrees of freedom (substrate + receptor one). The receptor produces a friction force that dissipates the kinetic energy of the c.o.m. of the substrate. To avoid this problem that would trap the substrate in a region of the receptor surface, both
The method has been successfully applied to study the docking of a phosphocholine molecule to the immunoglobulin McPC603 binding site in vacuo and in water.

Spherical boundary used for the MDD simulations. The atoms in the volume between the red and gray circle have position restraints.

Details of the McPC603 binding site.

The video of the MDD simulation of phosphocholine binding to the McPC603 is available Google+ site of the author.
The combination of this simple approach with essential dynamics sampling could in principle speed up docking processes where conformational changes in the receptor (or in the ligand) play a major role in the recognition.
BIBLIOGRAPHY
- Mangoni, D. Roccatano, A. Di Nola. Docking of flexible ligands to flexible receptors in solution by molecular dynamics simulations. Proteins: Struct., Funct., Genet., 35, 153-162 (1999).
- Di Nola, D. Roccatano, H. J. C. Berendsen. Molecular dynamics simulation of the docking of substrates to proteins. Proteins: Struct., Funct., Genet., 19, 174-182 (1994).

