At quite uncertain times and places,
The atoms left their heavenly path,
And by fortuitous embraces,
Engendered all that being hath.
And though they seem to cling together,
And form ‘associations’ here,
Yet, soon or late, they burst their tether,
And through the depths of space career.
James Clerk MaxwellFrom ‘Molecular Evolution’, Nature, 8, 1873. In Lewis Campbell and William Garnett, The Life of James Clerk Maxwell (1882), 637.
Molecular forces are originated by the interactions of the electronic clouds of the atoms in the molecular systems. A full treatment of these interactions also accounting for the dynamics of the nuclei requires the solution of the time-dependent Schroedinger equation (the top of the modeling pyramid). This approach would provide a more accurate physical representation of the behavior of the systems in time. However, as pointed before, nowadays this approach is impracticable due to the enormous amount of computer resources need to accomplish this task even for relatively small peptides in water systems. The solution to this impasse is the application of the so-called lex parsimoniae or Ockham’s razor, a powerful approach in problem-solving to get rid of the redundant complexity. In this case, the law of parsimony suggests changing the level of scale and account of the hidden degree of freedom using an effective or mean field potential.
Force fields
As mentioned in the introduction of the chapter, the atomic interactions in the classical MD are treated using analytic functions. The sum of these functions, usually referred to as force field function or effective potential, 



an example of force field, used in many programs for MD simulations, is the following:

Bonded interactions
The bonded interactions are usually described using functions depending on the coordinates of 2 to 4 particles. These interactions generally describe harmonic motions with force constants obtained from experimental crystallographic and spectroscopic data.
Bond vibrations
The first term (


![V_M(b)=\sum_{legami} D_e \left\{e^{[ - k_b(b-b_0) ]} -1 \right\}^2](https://s0.wp.com/latex.php?latex=V_M%28b%29%3D%5Csum_%7Blegami%7D+D_e+%5Cleft%5C%7Be%5E%7B%5B+-+k_b%28b-b_0%29+%5D%7D+-1+%5Cright%5C%7D%5E2&bg=ffffff&fg=444444&s=0&c=20201002)
where the parameter 

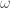
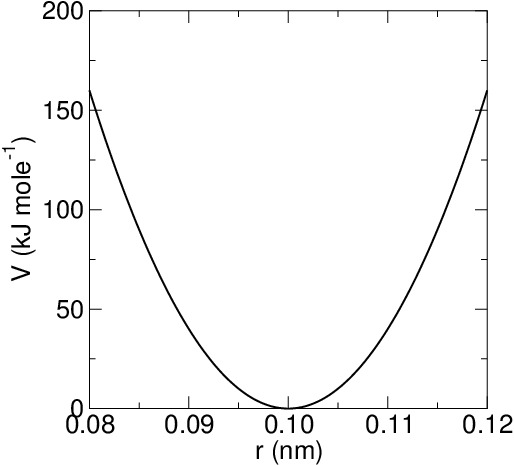
Figura 1: Examples of bond vibration potential: harmonic type (on the left); Morse type (on the right).
Bond angle vibration
The second term in force field Equation describes the interaction energy due to the deformation of the bond angle. This is a three-bodies interaction term since it depends on three atoms. The function represents harmonic angular vibrations, where $\theta_0$ is the reference value of the bond angle and 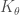
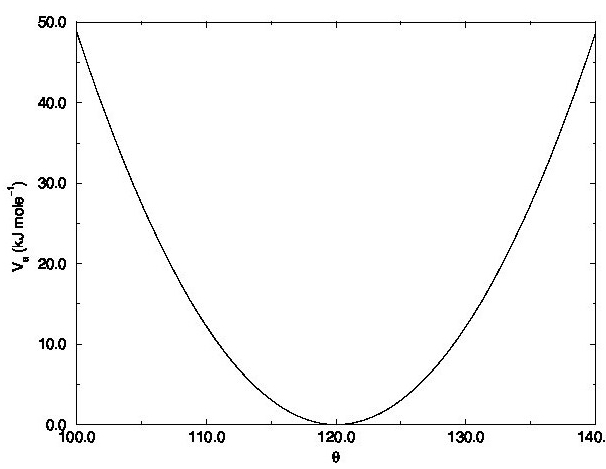
Figure 2: Example of bond angle vibration harmonic potential.
In some force fields, mixed bond-angle terms of type ![K_{b\theta}[b-b_0][\theta-\theta_0]](https://s0.wp.com/latex.php?latex=K_%7Bb%5Ctheta%7D%5Bb-b_0%5D%5B%5Ctheta-%5Ctheta_0%5D&bg=ffffff&fg=444444&s=0&c=20201002)
Four body interactions
In the Equ. 1, two terms are used to describe the following four-bodies interactions.
Improper dihedral vibrations
The first one describes the so-called improper dihedral angle vibrations. This term is used in order to account for the deformation of tetrahedral and planar geometries. The need to maintain tetrahedral geometries is due to the fact that in some force fields, apolar aliphatic hydrogens are not considered explicitly, but their effect is included by modifying the interaction functions of the atoms to which they are bound. In this way, chiral aliphatic carbons have only three coordinating atoms, this involves that during the simulation it can happen that these invert their chirality. Using improper dihedrals, this effect can be avoided by introducing an extra potential that keeps in place the right stereochemistry. The improper dihedral angle, i-j-k-l, is defined as the angle between the plane that is spanned by atoms i, j, and k and the plan spanned by atoms j, k, and l.
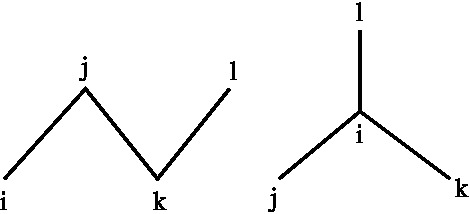
Figure 3: Definitions of improper dihedrals.
Torsion potentials
The other term for the four atom interactions describes the energy involved in the rotation around bonds. It is usually described as a sinusoidal function where 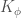
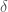

Figure 4: Example of torsion potential.
Derivation of the bond interaction parameters
In order to determine the parameters for the force fields, two paths are usually followed.
The most elegant method is to optimize these parameters by fitting to an energy function derived from quantum-mechanical calculations on small molecules. Bond and bond-angle can be obtained by normal mode analysis, while the dihedral term can be acquired by conformational analysis of the molecule around the specific bond. Alternatively, it is possible to optimize the force field parameters using experimental data: crystallographic structures, infrared and NMR spectroscopy, dynamic and thermodynamic properties of liquids as density and enthalpy of vaporization, free energy of solvation, just to mention some [Jorgensen83, Lifson83, Giglio69]. Using this approach, the model obtained is able to reproduce the experimental macroscopic properties of bulk liquids.
Non-bonded interactions
The last terms in Equ.~\ref{equ:forcef} describe the interactions between non-bonded atoms. The two terms are usually modeled using pair interaction functions describing van der Waals and Coulombic interactions of the atom pairs i and j having partial charges 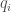


Van der Waals interactions
The short-range interactions are usually described using Lennard-Jones functions. The Lennard-Jones potential is composed of two terms that describe a repulsive and an attractive type interaction:
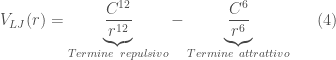


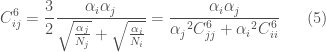
The attractive term in Equ.~\ref{lennardjones} describes the contribution of the non-bonded interactions of the so-called dispersive or London forces [London30, Israelachvili85]. The values of the constants 
the equation of Slater-Kirkwood [Margenau69]:
in which N represents the effective atomic electron number and 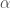


NOTE: The equation of state for a real gas can be expressed using the Virial formula

where $B$ and $C$ are the second and third coefficients of the Virial, respectively. For a good description of a gas in ideal conditions (very low dilution) it is already sufficient to consider only the $B$ coefficient. Applying the kinetic theory of gases, Rayleigh has found an analytic expression that connects $B$ to the atomic interaction function:

where r is the distance between the interacting molecule and the reference one. By replacing in V(r) the expression of the Lennard-Jones equation, it is possible to obtain an expression that allows determining the parameters 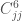
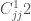
In the original form of the Lennard-Jones equation, the 


![V_{LJ} = 4 \epsilon \left[\left(\frac{\sigma}{r}\right)^{12} - \left(\frac{\sigma}{r}\right)^{6}\right]](https://s0.wp.com/latex.php?latex=V_%7BLJ%7D+%3D+4+%5Cepsilon+%5Cleft%5B%5Cleft%28%5Cfrac%7B%5Csigma%7D%7Br%7D%5Cright%29%5E%7B12%7D+-+%5Cleft%28%5Cfrac%7B%5Csigma%7D%7Br%7D%5Cright%29%5E%7B6%7D%5Cright%5D+&bg=ffffff&fg=444444&s=0&c=20201002)
The values of
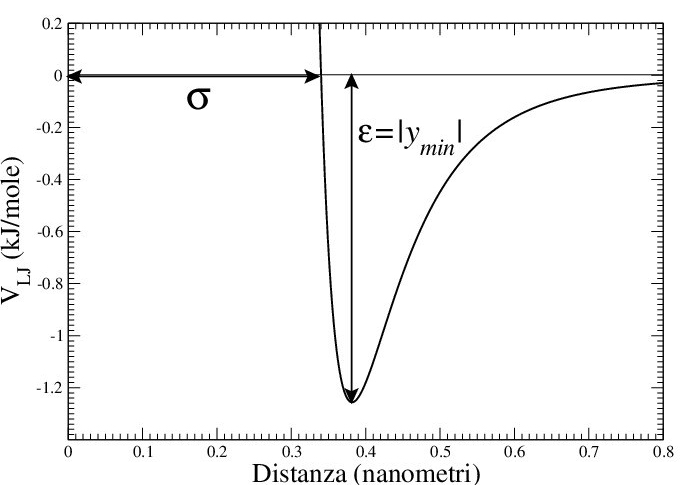
Figure 5: Example of Lennard-Jones potential.
Various types of combination rules exist, in one of the most widely used the new parameters are obtained in the following way:


The interactions between atoms covalently bound or not separate from each other by more than two bonds normally are not calculated. On the contrary, in the case of interactions between atoms separated by three bonds, the parameters of the LJ functions are properly reduced in order to avoid strong repulsions caused by the distances being too short. The parameters 
Electrostatic interactions
The electrostatic interactions are generally described using a coulomb term. Every atom of the system is provided with a partial atomic charge calculated using a quantum-mechanical method. The choice of the relative dielectric constant, $\varepsilon_r$, is still a matter of discussion in case one wants to simulate non-homogenous molecular systems, as proteins in water. Different values have been used that vary from 


where N is the atom number of the system, 
are the dipole moment and the induced dipole moment on the ith atom, respectively. Finally, the polarizability tensor is defined as follow:
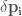

The previous equations set a system of 3N equations of 3N unknowns (each for the x, y, z components of every atom) that can be numerically solved. Another different way to introduce the polarizability in the simulated system is the use of the dummy charged particle, localized on each atom and bonded together using harmonic springs with an opportune force constant (Drude’s harmonic oscillators). The deformation of the spring resulting from the interactions with other atoms, produce a charge separation of the atoms and produce an induced dipole.
The hydrogen bond
Another important molecular interaction is the hydrogen bond. The term \em hydrogen bond \em is used in order to indicate all those atomic contacts in which the internuclear distances, 
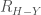
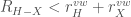


usually multiplied by a function of the angular orientation of the atoms involved.
Special interaction potentials
It is possible to add special terms to the force field potential function that describe the effect of external forces, that restrain the dynamics of the systems. In the case of proteins
or nucleic acids, these restraints confine the conformational space accessible to the molecule based experimental data. When these data are from X-ray diffraction or NMR, or other structural spectroscopic techniques, these potential restraints allow refining the molecular structure. Two examples of these potentials are reported in the following paragraphs.
Positional restraints
In case you want to keep a set of atoms vibrating around fixed positions in space, then
it is possible to define a harmonic potential that restraints the atoms to vibrate around a reference position 

This potential restraint is useful for simulations of solid surfaces, or to avoid structural deformation of large macromolecules during the equilibration of the surrounding solvent.
Restraint potential based on the crystallographic structure factors
The x-ray diffraction measurements provided the so-called structure factors, 

![V_{X}= \frac{1}{2}K_{X} \sum_{hkl}\left[F_{calc}(hkl)-F_{obs}(hkl)\right]^2](https://s0.wp.com/latex.php?latex=V_%7BX%7D%3D+%5Cfrac%7B1%7D%7B2%7DK_%7BX%7D+%5Csum_%7Bhkl%7D%5Cleft%5BF_%7Bcalc%7D%28hkl%29-F_%7Bobs%7D%28hkl%29%5Cright%5D%5E2&bg=ffffff&fg=444444&s=0&c=20201002)
where 
Distance dependent restraint potentials
The NMR spectroscopy provides accurate data on the average inter-protonic distance (Overhauser effect), on the dihedral angles (J coupling constants) and on the chemical shift that can be used to add precious structural information to the dynamical behavior of a macromolecule in an MD simulation. In this way, it is possible to obtain molecular structures that fulfill the experimental requirements. The NOE (Nuclear Overhauser Effect) intensities from NMR experiments can be converted in a set of superior limit distances 
![V({\bf{r}})=\frac{1}{2} \sum_{NOE}\left[max(0,<r_{ij}^{-1/3}>-r_{ij}^{ub})\right]^2](https://s0.wp.com/latex.php?latex=V%28%7B%5Cbf%7Br%7D%7D%29%3D%5Cfrac%7B1%7D%7B2%7D+%5Csum_%7BNOE%7D%5Cleft%5Bmax%280%2C%3Cr_%7Bij%7D%5E%7B-1%2F3%7D%3E-r_%7Bij%7D%5E%7Bub%7D%29%5Cright%5D%5E2&bg=ffffff&fg=444444&s=0&c=20201002)
where the function MAX provide the max value between the two arguments. The values of the coupling constant $J$ can be included in the force field using the relation [Torda93]:
![V_{J}({\bf{r}})= \frac {1}{2} K_{J} \sum_{\phi_i} \left[ <J(\phi_i({\bf{r}})> - J_i^{spe}\right]^2](https://s0.wp.com/latex.php?latex=V_%7BJ%7D%28%7B%5Cbf%7Br%7D%7D%29%3D+%5Cfrac+%7B1%7D%7B2%7D+K_%7BJ%7D+%5Csum_%7B%5Cphi_i%7D+%5Cleft%5B+%3CJ%28%5Cphi_i%28%7B%5Cbf%7Br%7D%7D%29%3E+-+J_i%5E%7Bspe%7D%5Cright%5D%5E2&bg=ffffff&fg=444444&s=0&c=20201002)
The coupling constant J depends on the torsion angle 
Finally, the chemical shifts 
![V_{\sigma}({\bf{r}})= \frac {1}{2} K_{\sigma} \sum_{i} \left[ <\sigma_i({\bf{r}})> - \sigma_i^{spe}\right]^2](https://s0.wp.com/latex.php?latex=V_%7B%5Csigma%7D%28%7B%5Cbf%7Br%7D%7D%29%3D+%5Cfrac+%7B1%7D%7B2%7D+K_%7B%5Csigma%7D+%5Csum_%7Bi%7D+%5Cleft%5B+%3C%5Csigma_i%28%7B%5Cbf%7Br%7D%7D%29%3E+-+%5Csigma_i%5E%7Bspe%7D%5Cright%5D%5E2&bg=ffffff&fg=444444&s=0&c=20201002)
where the sum is calculated over all the constrained atoms. The average 
Optimization of the molecular geometry
The potential function, described in the previous paragraphs, is used in molecular mechanics to explore the conformational energy surface of small molecules. This exploration would allow searching for those conformations that are energetically localized in minimum points. An exhaustive search procedure is difficult to perform for molecules having many dihedrals. However, for the optimization of the structure to the closest minimum, it is important to eliminate possible contacts in the starting conformation of the MD simulations. In fact, incorrect molecular conformation can create problems in the integration algorithms of the equation of motion.
A molecular configuration is in a conformational energy minimum if the following condition is verified:

where N is the total number of atoms of the system, 
Among the methods used to search for local minima the most widely used are:
- The steepest descent method
- The conjugate gradient method
In the first case, given a molecular geometry, defined by the coordinates 
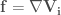
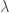


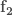

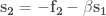
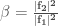
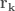

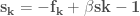
with 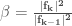
READING LIST
- D. Roccatano. A short Introduction to the Molecular Dynamics Simulation of Nanomaterials. Book chapter in Micro and Nanomanofacturing, second edition. Editors: W. Ahmed and M. J. Jackson. 2017. Springer.
- Roccatano, D., Theoretical Study of Nanostructured Biopolymers Using Molecular Dynamics Simulations: A Practical Introduction, in Nanostructured Soft Matter. 2007, Springer. p. 555-585.
- Roccatano, D., Computer Simulations of Biomolecules in Non-Aqueous and Semi- Aqueous Solvent Conditions, in Advances in Protein and Peptide Sciences, B.M. Dunn, Editor. 2013, Bentham Science Publishers. p. 318.
- Roccatano, D., The molecular dynamics simulation of peptides on gold nanosurfaces. Nanoparticles in Biology and Medicine: Methods and Protocols, 2020: p. 177-197.
- van Gunsteren, W.F. and H.J.C. Berendsen, Computer simulation of molecular dynamics: Methodology, Applications, and perspectives. Angew. Chem. Int. Eng. Ed., 1990. 29: p. 992-1023.
- Leach, A.R., Molecular Modelling. Principles and Applications. 2 ed. 2001: Prentice Hall.
- Allen, M.P. and D.J. Tildesley, Computer Simulation of Liquids. 1989: Oxford University Press.
- Berendsen, H.J.C., Simulating the physical world: hierarchical modeling from quantum mechanics to fluid dynamics. 2007, Cambridge ; New York: Cambridge University Press.


Pingback: Calculus in a Nutshell: functions and derivatives | Danilo Roccatano
This is so important! Are there any resources you can recommend for learning more about biomolecular interactions?
https://depixus.com/introducing-technology/
LikeLike
Thank you for your kind words. I’ve updated my article to include references to my review publications and additional relevant books. However, I must acknowledge that there are numerous excellent articles available on the topic of MD simulations. Nonetheless, it seems that your company (Depixus) is producing exceptional machines that are crucial for validating or enhancing models used in MD simulations!
LikeLike