My blog in italian on this topics is very popular and for this reason I decided to add an English translation (when I have some free time, I will also translate the text in the Figure and Table). So be tune and more will come!
The stability of a crystal lattice at constant T and P conditions is linked to the Gibbs free energy of lattice formation by the relations


If 

The lattice formation process starting from an ion in gas phase is so exothermic that at room temperature (298 K) the entropic contribution can be neglected (the entropic effect due to temperature becomes completely negligible only at temperatures close to absolute zero, 
The enthalpy of dissociation of the crystal lattice (or reticular enthalpy, 


The dissociation of the crystal lattice is endothermic, therefore the values of $ latex {\Delta H_c} $ are always positive. The opposite reaction or the formation of the crystal

it is highly exothermic and therefore the enthalpy value is negative.
The value of
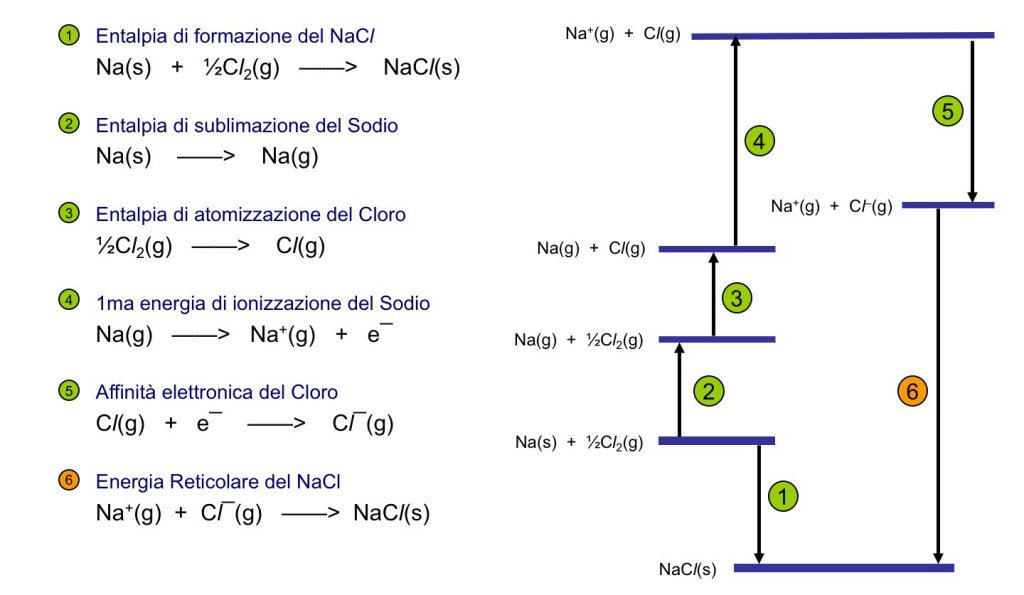

Since enthalpy is a function of state, by the law of Hess the sum of the enthalpy variation along a complete cycle must be zero:
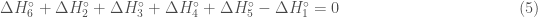
Therefore, the sum of the enthalpies is given by 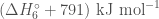
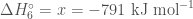

Table 2 lists the reactions for the calculation of the energy of formation of the 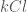

In this case, the sum of the enthalpies is given by 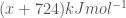


As a last example, in Figure 2, the cycle for the 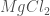
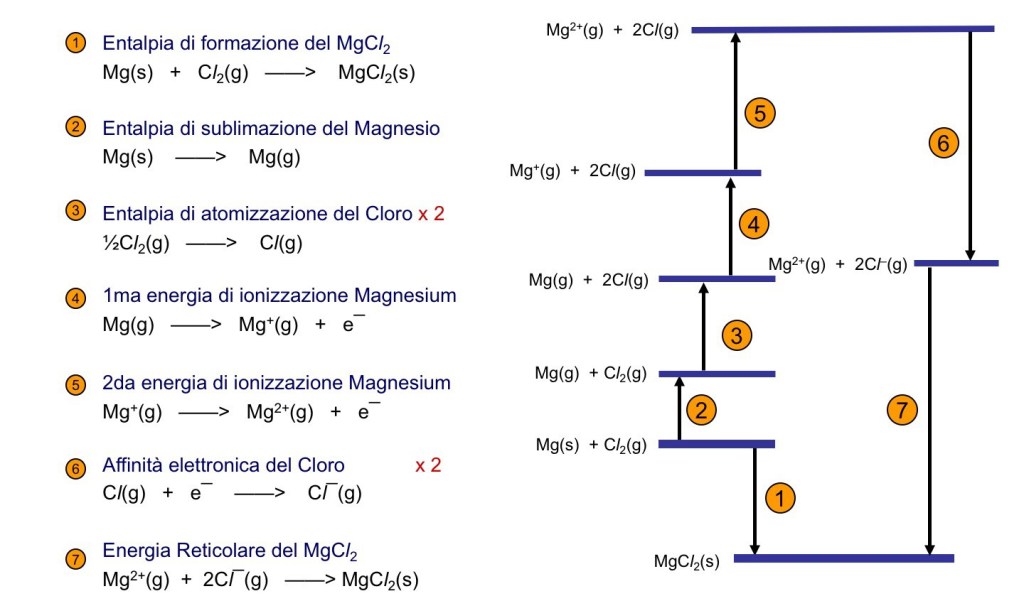 .
.The reticular enthalpy allows to evaluate the nature of the bond present in the solid. If the value calculated with the ionic model agrees well with the experimental one, as in this example, it is a reasonable approximation to adopt the ionic model for the compound. Any discrepancy become significant when there is an amount of covalence in the bonds.
To calculate the reticular enthalpy of an ionic compound it is necessary to consider the different contributions to its energy, such as:
- The electrostatic attractions and repulsions of the ions;
- The dispersion interactions: for ions of limited polarizability this contribution is only about 1% of the electrostatic contribution.
- Repulsive forces due to the overlap of electronic distributions. Repulsive interactions between ions are essential to the stability of the ionic solid; in the absence of repulsive interactions, the cations and anions would unite, ie they would collapse. A
(when there is no thermal agitation and, therefore, there is no nuclear kinetic energy), the ions adopt distances such that the attractions compensate for the repulsive interactions.
- The kinetic energy deriving from the vibration motions of the ions can be neglected (except for the contribution of the zero point energy) if we consider the solid at absolute zero.
In Table 3 some values of 
An acceptable agreement with the experimental data means that the compound is ionic; an unsatisfactory agreement means that we are facing a significant degree of covalence.
TABLE 3: Measured and calculated reticular enthalpies..
| Composto |  |  | 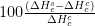 |
| (calcolato) | (sperimentale) | ||
| LiF | 1033 | 1037 | 0.4 |
| LiCl | 845 | 852 | 0.8 |
| LiBr | 798 | 815 | 2.1 |
| LiI | 740 | 761 | 2.8 |
| NaF | 915 | 926 | 1.2 |
| NaCl | 778 | 786 | 1.0 |
| NaBr | 739 | 752 | 1.7 |
| NaI | 692 | 705 | 1.8 |
| KF | 813 | 821 | 1.0 |
| KCl | 709 | 717 | 1.1 |
| KBr | 680 | 689 | 1.3 |
| KI | 640 | 649 | 1.4 |
| RbF | 778 | 789 | 1.4 |
| RbCl | 686 | 695 | 1.3 |
| RbBr | 659 | 668 | 1.3 |
| RbI | 622 | 632 | 1.6 |
| CsF | 748 | 750 | 0.3 |
| CsCl | 652 | 676 | 3.6 |
| CsBr | 632 | 654 | 3.4 |
| CsI | 601 | 620 | 3.1 |
| CuCl | 904 | 993 | 9.0 |
| CuBr | 870 | 976 | 10.9 |
| CuI | 833 | 963 | 13.5 |
| AgF | 920 | 969 | 5.1 |
| AgCl | 833 | 912 | 8.7 |
| AgBr | 816 | 900 | 9.3 |
| AgI | 778 | 886 | 12.2 |
| TlCl | 686 | 748 | 8.3 |
| TlBr | 665 | 732 | 9.2 |
| TlI | 636 | 707 | 10.0 |
The percentage difference (last column) between the experimental and calculated value clearly shows that the alkali metal halides show a good agreement when the halogen is poorly polarizable (like the fluoride ion, 

The worst difference is found with combinations of a cation and anion both very polarizable, such as CuI, AgI and TlI. These compounds are substantially covalent, and therefore


Pingback: CRYSTAL STRUCTURES |
Pingback: The calculation of the Madelung constant |
Pingback: Madelung常数的计算 |
Pingback: Madelung定数の計算 |
Pingback: Il calcolo della costante di Madelung |
Pingback: El cálculo de la constante de Madelung |