Understanding the mechanisms of the molecular recognition has fundamental impacts in medicine and biotechnology. It plays an important role in discovering new drugs and in developing new biocatalyst. The theoretical study of these mechanisms has boosted the development of approximated but fast methods for screening large compound libraries and protein-protein complexes. Continue reading
Research

Protein Engineering and Directed Evolution
Phytase


- A. Shivange, U. Schwaneberg, D. Roccatano. Conformational dynamics of active site loop in Escherichia coli phytase. Biopolymers. 93(11), 994–1002, (2010).
Monooxygenase P450 BM3
Figure.
- S. Wong, N. Wu, D. Roccatano, M. Zacharias and U. Schwaneberg. Sensitive assay for laboratory evolution of hydroxylases toward aromatic and heterocyclic compounds. J. Biomol. Screen, 10, 246-252 (2005).
- Nazor, S. Dannennmann, R. Obeg Adjei, Y. B. Fordjour, T. I. Ghampson, M. Blanusa, D. Roccatano, U. Schwaneberg. Laboratory evolution of P450 BM3 for mediated electron transfer yields and activity-improved & reductase-independent variant. Protein Eng. Des. Sel., 21(1), 29-35, (2008).
- K. L. Tee, D. Roccatano, S. Stolte, J. Arning, B. Jastorff, U. Schwaneberg. Ionic liquids as cosolvents for biotransformation catalyzed by P450 BM-3. Green Chem., 10, 117-123, (2008).
Share this:

Studies of preferential solvation of peptides and proteins in solution
Chemical environment (pH, salts, co-solvents) plays an important role in the stabilization of secondary structure forming peptides and proteins in solution. The presence of co-solvent in aqueous solution can increase the structural stability as well as to promote denaturation or conformational changes. Continue reading
Share this:

Study of Structural and Dynamical Properties of Peptides in Solutions
The recent progress on time-resolved optical spectroscopy and FRET techniques have opened new exciting perspectives to the understanding the short timescale dynamics of peptides in solution. In fact, experimental data provided by these measurements can be directly compared with the results of computer simulations.
Continue readingShare this:

Adsorption mechanism of an antimicrobial peptide on carbonaceous surfaces: A molecular dynamics study
Danilo Roccatano, Edita SarukhanyanThe Journal of Chemical Physics 146, 074703 (2017); doi: http://dx.doi.org/10.1063/1.4975689
 Cover Page
Cover PagePeptides are versatile molecules with applications spanning from biotechnology to nanomedicine. They exhibit a good capability to unbundle carbon nanotubes (CNT) by improving their solubility in water. Furthermore, they are a powerful drug delivery system since they can easily be uptake by living cells, and their high surface to volume ratio facilitates the adsorption of molecules of different nature. Therefore, understanding the interaction mechanism between peptides and CNT is important for designing novel therapeutically agents. In this paper, the mechanisms of the adsorption of antimicrobial peptide Cecropin A – Magainin 2 (CA-MA) on a graphene nanosheet (GNS) and on an ultra-short single-walled CNT are characterized using molecular dynamics simulations. The results show that the peptide coats both GNS and CNT surface through preferential contacts with aromatic side chains. The peptide packs compactly on the carbon surfaces where the polar and functionalize Lys side chains protrude into the bulk solvent. It is shown that the adsorption is strongly correlated to a loss of the peptide helical structure. In the case of the CNT, the outer surface is significantly more accessible for adsorption. Nevertheless when the outer surface is already covered by other peptides, a spontaneous diffusion, via the amidated C-terminus, into the interior of the CNT was observed within 150 ns of simulation time. We found that this spontaneous insertion into the CNT interior can be controlled by the polarity of the entrance rim. For the positively charged CA-MA peptide studied, hydrogenated and fluorinated rims, respectively, hinder and promote the insertion.

Share this:

Molecular Machines within us: Citrate Synthase a Pac-Enzyme
Citrate Synthase (CS) is an enzyme localized in the mitochondria of our cells where it plays an important role in the aerobic respiration cycle by transforming oxaloacetate molecules (on the right side of the picture) in citrate (on the top left side) with the assistance of the acetyl-coenzyme A (CoA) molecule. Continue reading
Share this:

Exploring the Molecular Machines within us: A Fantastic Voyage
“To see a World in a Grain of Sand
And a Heaven in a Wild Flower,
Hold Infinity in the palm of your hand
And Eternity in an hour.”
―William Blake, Auguries of Innocence.
INTRODUCTION
This blog is based on public talks that I have delivered at the University of Lincon, UK and at the Gravity Fields Festival 2016. Here I give a short summary of talk topics.
Nature is an unlimited source of great inspiration (and imitation) for scientist and engineering. In fact, the continuous advance in the knowledge of the complex machinery of life is producing profound impacts in the modern societies. Life, in the form that we know, definitively exploited what we now call “nanotechnology” to emerge. Living cells are crowded with fascinating molecular machines with a large variety of functions not yet completely explored. Nature as a blind and patient engineer builds these machines without a defined blueprint but utilizing the power of the evolution.
Continue readingShare this:

Molecular Properties of Astaxanthin in Water/Ethanol Solutions from Computer Simulations
Khadga Jung Karki, Susruta Samanta, and Danilo Roccatano*
J. Phys. Chem. B, August 2016
Astaxanthin (AXT) is a reference model of xanthophyll carotenoids, which is used in medicine and food industry, and has potential applications in nanotechnology. Because of its importance, there is a great interest in understanding its molecular properties and aggregation mechanism in water and mixed solvents. In this paper, we report a novel model of AXT for molecular dynamics simulation. Continue reading
Share this:

2D phase Transition of Simple Liquids: Methanol on Graphene
In a recent paper [1], we have studied the phase transitions of monolayer of methanol molecules confined between two graphene sheets.

Structural order emerging in the liquid state necessitates a critical degree of anisotropy of the molecules. For example, liquid crystals and Langmuir monolayers require rod/disc-shaped and long chain amphiphilic molecules, respectively, to break the isotropic symmetry of liquids.
In the paper, we have presented results from molecular dynamics simulations demonstrating that in two-dimensional liquids, a significantly smaller degree of anisotropy is sufficient to allow structural organization. In fact, the condensed phase of the smallest amphiphilic molecule, methanol, confined between two or adsorbed on, graphene sheets form a monolayer characterized by long chains of molecules. Intra-chain interactions are dominated by hydrogen bonds, whereas inter-chain interactions are dispersive. Upon a decrease in density toward a gas-like state, these strings are transformed into rings. The two-dimensional liquid phase of methanol undergoes another transition upon cooling; in this case, the order-disorder transition is characterized by a low-temperature phase in which the hydrogen bond dipoles of neighboring strings adopt anti-parallel orientation.
REFERENCE
- R. Zangi and D. Roccatano. Strings-to-Rings Transition and Anti-parallel Dipole Alignment in Two-Dimensional Methanols. Nano Lett. 16, 5, 3142-3147, (2016). DOI: 10.1021/acs.nanolett.6b00460
Share this:

Invited Seminar at Norwich
On 3rd February 2016, Danilo Roccatano visited the School of Computing Science of the University of East Anglia in Norwich hosted by Dr Steven Hayward. He gave the invited seminar:
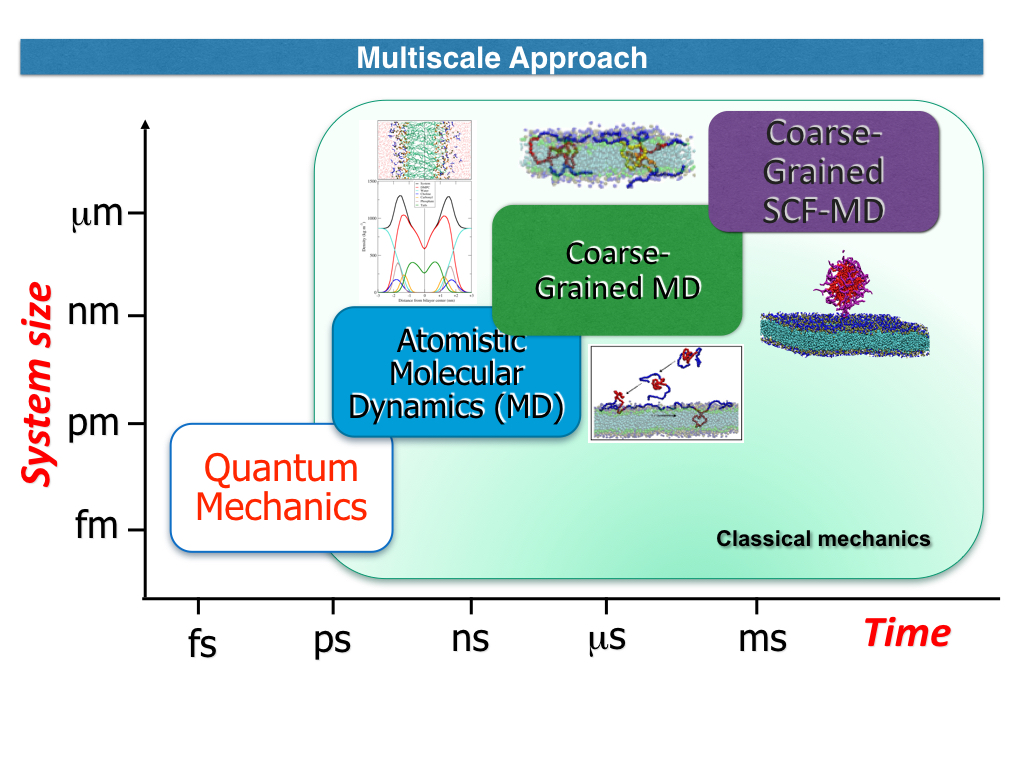
Study of Interaction Mechanisms of Block Copolymers with Biological Interfaces
Abstract
Polyethylene oxide and polypropylene oxide homopolymers as well as block copolymers based on them (Poloxamers or Pluronics®) have many applications in biotechnology and in pharmacology. This versatility is due to their biocompatibility and tuneable properties. Still the molecular mechanisms of their interactions with biological systems remain not fully investigated. A powerful and versatile approach to study these processes is the Molecular Dynamics (MD) simulation method that allows exploring these systems on scale of different order of magnitude in length and time. In the last years, we have developed for these purpose full atoms and coarse-grained models of these polymers that have been successfully tested against several experimental data in solution, and at interface with lipid bilayers. Using a recently proposed and developed Self Consistent density Field MD method, we also accomplished to perform large-scale simulations study of polymeric micelles formation and their interaction with lipid bilayers. These results have unrevealed possible mechanisms of single polymer and micelle interaction with lipid bilayers. In this talk, I will summarize the main achievements and future directions of these studies.