Danilo Roccatano, Edita SarukhanyanThe Journal of Chemical Physics 146, 074703 (2017); doi: http://dx.doi.org/10.1063/1.4975689
 Cover Page
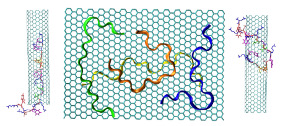
Cover PagePeptides are versatile molecules with applications spanning from biotechnology to nanomedicine. They exhibit a good capability to unbundle carbon nanotubes (CNT) by improving their solubility in water. Furthermore, they are a powerful drug delivery system since they can easily be uptake by living cells, and their high surface to volume ratio facilitates the adsorption of molecules of different nature. Therefore, understanding the interaction mechanism between peptides and CNT is important for designing novel therapeutically agents. In this paper, the mechanisms of the adsorption of antimicrobial peptide Cecropin A – Magainin 2 (CA-MA) on a graphene nanosheet (GNS) and on an ultra-short single-walled CNT are characterized using molecular dynamics simulations. The results show that the peptide coats both GNS and CNT surface through preferential contacts with aromatic side chains. The peptide packs compactly on the carbon surfaces where the polar and functionalize Lys side chains protrude into the bulk solvent. It is shown that the adsorption is strongly correlated to a loss of the peptide helical structure. In the case of the CNT, the outer surface is significantly more accessible for adsorption. Nevertheless when the outer surface is already covered by other peptides, a spontaneous diffusion, via the amidated C-terminus, into the interior of the CNT was observed within 150 ns of simulation time. We found that this spontaneous insertion into the CNT interior can be controlled by the polarity of the entrance rim. For the positively charged CA-MA peptide studied, hydrogenated and fluorinated rims, respectively, hinder and promote the insertion.