The citrate synthase is a model enzyme for the study of the dynamics of domain motions. It is a key protein in the metabolic cycle of the cell (Krebs cycle) as it catalyzes the Claisen condensation of acetyl-coenzyme A with oxaloacetate, to form citrate and coenzyme A. The catalytic mechanism is very complex with many unknowns. In most organisms its structure comprises two identical subunits. Continue reading
Author: Danilo Roccatano

Molecular Machines within us: The Glutamate Synthase
Glutamate Synthases are key enzymes in the early stages of the assimilation of ammonia in bacteria, yeast, and plants. Azospirillum brasilense glutamate synthase (GltS) is a complex iron-sulfur flavoprotein that catalyzes the reductive transfer of L-glutamine amide group to the (C2) carbon of 2-oxoglutarate(2OG) yielding two molecules of L-glutamate and whose function requires the transfer of ammonia and electrons among distinct catalytic sites. Continue reading
Share this:

Molecular Machines within us: Folding/Unfolding Mechanism of the Cytochrome c
The Cytochrome c (Cytc) is a small heme protein of ~100 amino acids. It presents in the mitochondrion of eukaryotic cells and it is loosely associated with their inner membrane. Cytc is highly water-soluble and is an essential component of the electron transport chain in mitochondria where it carries one electron. Continue reading
Share this:

Fluorinated solvents models for MD simulations
Experimental studies on peptides and proteins stability and folding in solution are usually made in the presence of membrane mimic nonaqueous solvents. These conditions facilitate the study of hydrophobic stabilization effects.
Continue readingShare this:

Secondary structure forming peptides in solution: ß-turns, ß-hairpins and three-stranded peptides
sMy research on peptides is focused on those that are able to form secondary structure elements in solution like 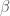
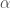
Share this:

Effect of the fluorinated solvents on the stability of secondary structure forming peptides
The effect of 2,2,2-trifluoroethanol (TFE) as a cosolvent on the stability of three different secondary structure-forming peptides, the Melittin, the Betanova, and the β-hairpin II from the bacterial protein GB1 (see the crystal structures in Figure 1), was studied using molecular dynamics simulations. Continue reading
Share this:

Study of the conformational transition in amyloidogenic peptides
Understanding the conformational transitions that trigger the aggregation and amyloidogenesis of otherwise soluble peptides at atomic resolution is of fundamental relevance for the design of effective therapeutic agents against amyloid-related disorders. Continue reading
Share this:

Application of Quasi-Gaussian entropy theory to predict the properties of simple fluids and biomolecules in solution
The Quasi-Gaussian Entropy theory (QGE) is a new statistical mechanics theory (Amadei et al., J. Chem. Phys. (1996), 104, 1560-1574), that can be used to predict the physical-chemistry properties of real and simulated systems in a very wide temperature range. A new expression of the Clausius-Clapeyron equation, based on the QGE, for the evaluation of the liquid-vapor equilibrium pressure of pure liquids, was developed [1]. The new equation is able to predict the liquid-vapor equilibrium pressure curve, with high accuracy, over a large temperature range and for different fluids like water, methanol, and mercury.
Continue readingShare this:

Molecular Dynamics Simulations Combined to X-Ray Spectroscopies XANES and EXAFS
In Laurea thesis (equivalent to M.Sc. Diploma Thesis, discussed in 1992), I have for the first time investigated and attempted the combination Molecular Dynamics (MD) simulations and theoretical spectra calculation for the interpretation of experimental XANES (X-ray Absorption Near-Edge Spectroscopy) [4]. The method was used to study the aquatrisimidazole copper (II) sulfate complex’s crystal structure, but the results not yet published. The thesis is in Italian, and it is available on request to the interested reader.
Continue readingShare this:

Parallel computing and molecular dynamics simulations
The recent technical developments in parallel computing have made available large parallel computing facilities at relatively contained costs that have stimulated an intense developing activity to realize efficient parallel programs for the scientific and technical calculation. One of the scientific fields that most benefit from these developments is computational chemistry and in particular Molecular Dynamics (MD). The reason of that is related to the increasing interest in the study, with this technique, larger molecular systems for long simulation times. Continue reading

