In Laurea thesis (equivalent to M.Sc. Diploma Thesis, discussed in 1992), I have for the first time investigated and attempted the combination Molecular Dynamics (MD) simulations and theoretical spectra calculation for the interpretation of experimental XANES (X-ray Absorption Near-Edge Spectroscopy) [4]. The method was used to study the aquatrisimidazole copper (II) sulfate complex’s crystal structure, but the results not yet published. The thesis is in Italian, and it is available on request to the interested reader.
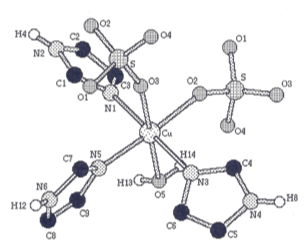
Figure 2: Crystal structure of the aquatrisimidazole copper (II) sulfate complex.
Subsequently, we have also applied a novel approach, to combining MD simulations to refine experimental data from Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy on the analysis of different molecular systems [1,2,3].
The approach is schematized in the following diagram.

First, MD (or also Montecarlo, MC) simulations are used to obtain a pair distribution functions model between a central photo-absorber and its first neighbours scattering atoms. The distributions functions are then used as a starting model to calculate the EXAFS spectra. By comparing the calculate spectra with the experimental EXAFS signal is possible to refine the accuracy of the pair distribution functions and obtain accurate structural data such as the coordination number and the distance of the scattering atoms from the central one.
The procedure has the advantage that can be used to accurately validate and optimize intermolecular potential models used for the MD simulation. The method was used studying aqueous solutions of ions and small molecules [1,2,3]. Subsequently also extended to more complex systems like metalloproteins, inorganic complexes, and organometallic compounds. These studies were among the first applications of MD simulations for the interpretation and refinement of the EXAFS spectra of complex molecular systems.
REFERENCES
- D’Angelo, A. Di Nola, A. Filipponi, N. V. Pavel, D. Roccatano. An extended X-ray absorption fine structure study of aqueous solutions. J. Chem. Phys., 100, 985-994 (1994).
- D’Angelo, N. V. Pavel, H. F. Nolting, D. Roccatano. Multielectron excitations at the L-edges of barium in aqueous solution. Phys. Rev. B, 54, 12129-12138 (1996).
- Roccatano, H. J. C. Berendsen, P. D’Angelo. Assessment of the validity of intermolecular potential models used in molecular dynamics simulations by extended X-ray absorption fine structure spectroscopy: A case study of Sr in methanol solution. J. Chem. Phys., 108, 9487-9497 (1998).
- Roccatano. Determinazione della struttura dello Cu(II) aquatrisimidazolo solfato mediante dinamica molecolare e spettroscopia XANES. Tesi di Laurea, equivalent to a Master Thesis (in Italian). Advisors: Prof’s A. Di Nola and M. Barteri, Rome 1992.

