A box without hinges, key, or lid,
JRR Tolkien, The Hobbit
Yet golden treasure inside is hid.
Easter 2022 is at the door and the occasion for the traditional appointment to talk about eggs and their mathematical shapes. This year with the help of my sons, we have created the following Instructable for STEM education:
https://www.instructables.com/Modelling-and-Designing-of-Bird-Eggs-for-3D-Printi/
The project aims to show how to use a simple mathematical model to generate the 3D form of real bird eggs utilizing several parameters. The 3D egg models can be saved as an STL file and then printed using a 3D printer. The printed egg can be painted or modified with a CAD program to add functionalities for egg-based gadgets or toys. An example of a modification to create a LED decorated egg is explained in detail.
More recently for fun, I have published another one using the same approach:
https://www.instructables.com/The-Eggyrint/
The egg modelling topic has been covered in previous article, and the interested reader can complement the information in the Instructable with other information provided in the following articles:
https://wordpress.com/post/daniloroccatano.blog/3792
https://wordpress.com/post/daniloroccatano.blog/5171
https://wordpress.com/post/daniloroccatano.blog/6760
The Instructable gives the possibility to 3D print and modifies the 3D shape of bird eggs. It can be used for research, teaching and fun. I hope you will enjoy it, and constructive comments and suggestions are always welcome!
AUGURO A TUTTI I LETTORI UNA BUONA PASQUA E PACE IN TERRA
WÜNSCHT ALLEN LESERN FROHE OSTERN UND FRIEDEN AUF ERDEN
I WISH TO READER A HAPPY EASTER AND PEACE ON EARTH






 , where N is the total number of electrons in the system. It was clear that a reduction, using ad hoc approximations, of the description of the dynamic behaviour of atoms using a classic physics model would be necessary to overcome this problem. In the classical representation, the electrons on the atoms are not explicitly considered, but their mean-field effect is taken into account. Alder and Wainwright performed the first simulation of an atomic fluid using this approximation approximately 63 years ago (1957). They developed and used the method to study simple fluids by means of a model representing atoms as discs and rigid spheres. These first pioneer studies mark the birth of the classical molecular dynamics (MD) simulation technique. The successive use of more realistic interaction potentials has allowed obtaining simulations comparable to experimental data, showing that MD can be a valuable tool for surveying the microscopical properties of physical systems. The first simulations of this type were carried out by Rahman and Verlet (1964): in these simulations, a Lennard-Jones-type potential was used to describe the atomic interactions of argon in the liquid state. Another significant hallmark in this field was the simulation of the first protein (the bovine pancreatic trypsin inhibitor) by McCammon and Karplus in 1977. In the following years, the success obtained in reproducing structural properties of proteins and other macromolecules led to a great spread of the MD within structural biology studies. The continuous increase of computer power and improvement of programming languages has concurred with further refinement of the technique. Its application was progressively expanded to more complex biological systems comprising large protein complexes in a membrane environment. In this way, MD is becoming a powerful and flexible tool with applications in disparate fields, from structural biology to material science.
, where N is the total number of electrons in the system. It was clear that a reduction, using ad hoc approximations, of the description of the dynamic behaviour of atoms using a classic physics model would be necessary to overcome this problem. In the classical representation, the electrons on the atoms are not explicitly considered, but their mean-field effect is taken into account. Alder and Wainwright performed the first simulation of an atomic fluid using this approximation approximately 63 years ago (1957). They developed and used the method to study simple fluids by means of a model representing atoms as discs and rigid spheres. These first pioneer studies mark the birth of the classical molecular dynamics (MD) simulation technique. The successive use of more realistic interaction potentials has allowed obtaining simulations comparable to experimental data, showing that MD can be a valuable tool for surveying the microscopical properties of physical systems. The first simulations of this type were carried out by Rahman and Verlet (1964): in these simulations, a Lennard-Jones-type potential was used to describe the atomic interactions of argon in the liquid state. Another significant hallmark in this field was the simulation of the first protein (the bovine pancreatic trypsin inhibitor) by McCammon and Karplus in 1977. In the following years, the success obtained in reproducing structural properties of proteins and other macromolecules led to a great spread of the MD within structural biology studies. The continuous increase of computer power and improvement of programming languages has concurred with further refinement of the technique. Its application was progressively expanded to more complex biological systems comprising large protein complexes in a membrane environment. In this way, MD is becoming a powerful and flexible tool with applications in disparate fields, from structural biology to material science.


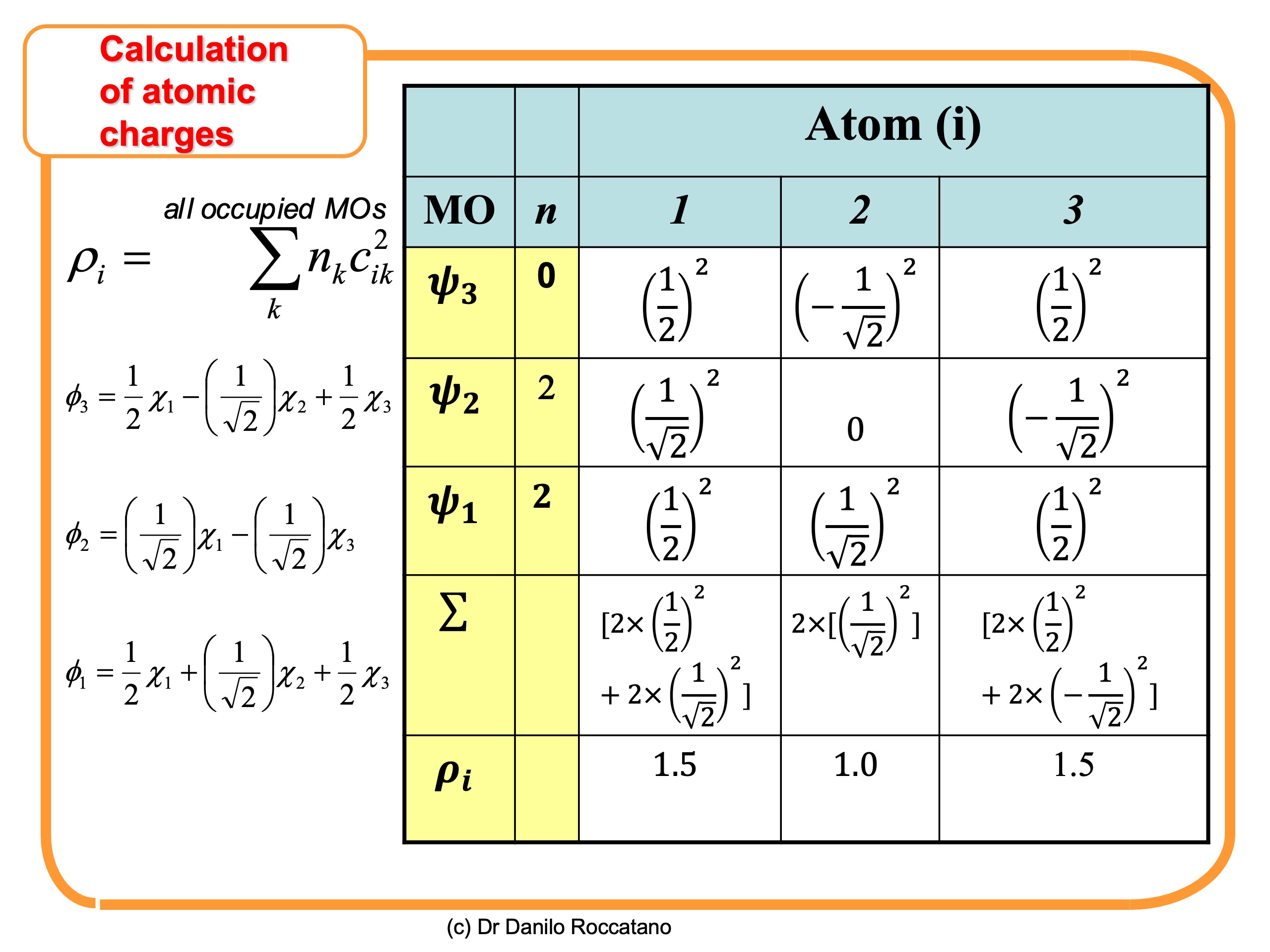
 (1)
(1) is the number of electrons in the
is the number of electrons in the 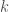 orbital.
orbital.
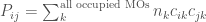
 of the allyl radical is reported. The result shows an equal contribution of 0.707 for both bonds.
of the allyl radical is reported. The result shows an equal contribution of 0.707 for both bonds.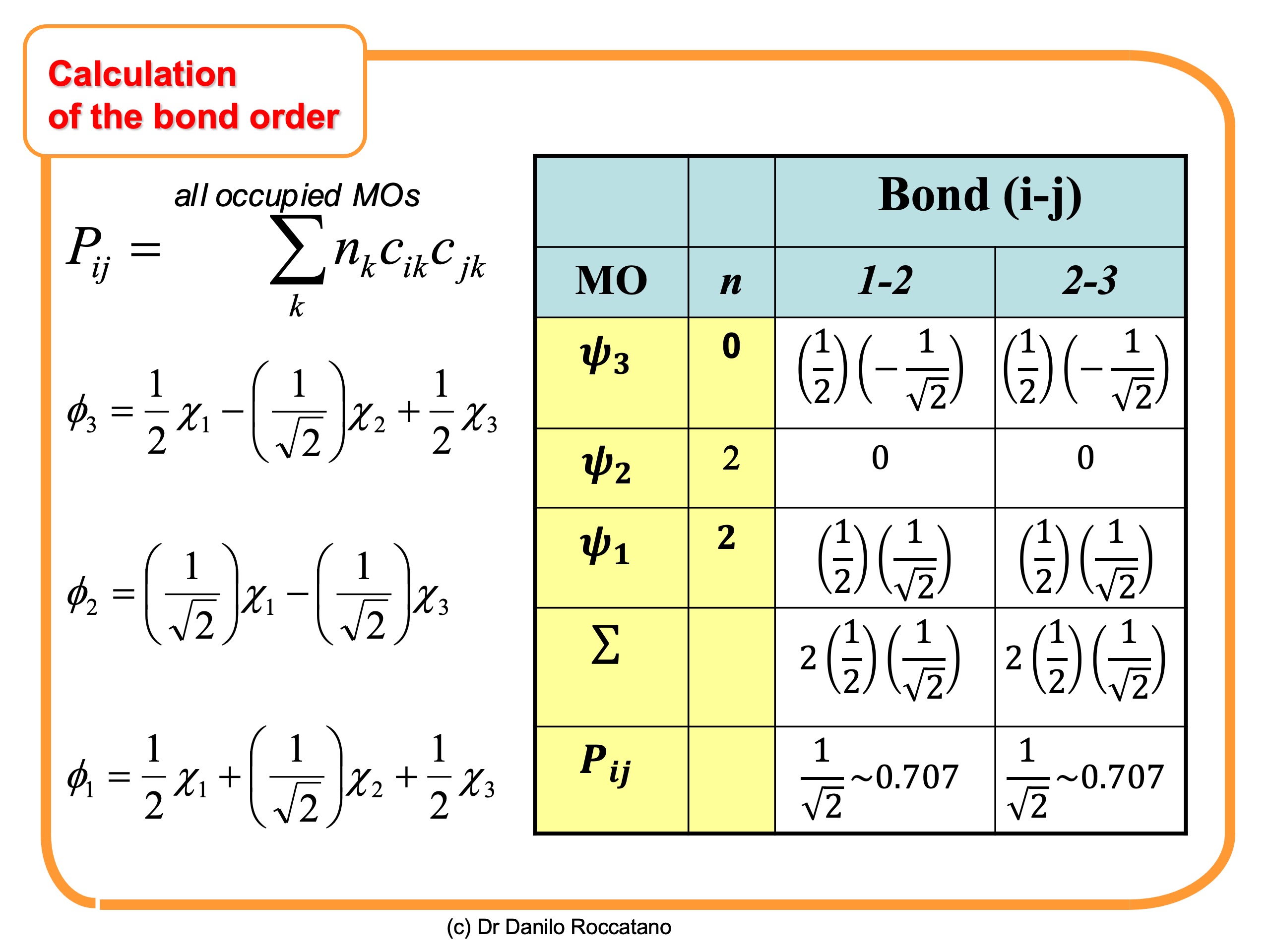
 as shown in the examples reported in the figure below.
as shown in the examples reported in the figure below.



 -bonds. In this third article, we will apply the method to cyclic molecules and will derive some other useful properties.
-bonds. In this third article, we will apply the method to cyclic molecules and will derive some other useful properties. 
