At quite uncertain times and places,
The atoms left their heavenly path,
And by fortuitous embraces,
Engendered all that being hath.
And though they seem to cling together,
And form ‘associations’ here,
Yet, soon or late, they burst their tether,
And through the depths of space career.
James Clerk MaxwellFrom ‘Molecular Evolution’, Nature, 8, 1873. In Lewis Campbell and William Garnett, The Life of James Clerk Maxwell (1882), 637.
Molecular forces are originated by the interactions of the electronic clouds of the atoms in the molecular systems. A full treatment of these interactions also accounting for the dynamics of the nuclei requires the solution of the time-dependent Schroedinger equation (the top of the modeling pyramid). This approach would provide a more accurate physical representation of the behavior of the systems in time. However, as pointed before, nowadays this approach is impracticable due to the enormous amount of computer resources need to accomplish this task even for relatively small peptides in water systems. The solution to this impasse is the application of the so-called lex parsimoniae or Ockham’s razor, a powerful approach in problem-solving to get rid of the redundant complexity. In this case, the law of parsimony suggests changing the level of scale and account of the hidden degree of freedom using an effective or mean field potential.
Continue reading


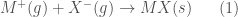
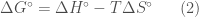
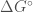 risulta più negativa per la formazione della struttura
risulta più negativa per la formazione della struttura 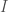 che per quella della struttura
che per quella della struttura 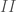 , la transizione
, la transizione  sarà spontanea e il solido avrà quella struttura.
sarà spontanea e il solido avrà quella struttura.






 . Questi potenziali di energia intermolecolari possono essere classificati in base al tipo di interazioni che descrivono.
. Questi potenziali di energia intermolecolari possono essere classificati in base al tipo di interazioni che descrivono. 

