Important structural components of proteins, like linker loops and intrinsically disordered regions, are highly flexible and constantly change shape in solution. These flexible protein regions — especially those containing glycine- and serine-rich segments — do not behave like neatly folded proteins. They fluctuate, breathe, and explore broad conformational landscapes. These motions can often be central to biological function. But capturing them consistently, both structurally and dynamically, remains challenging. To understand the physics of this flexibility, we often turn to short model peptides that isolate the essential ingredients of chain dynamics. In an earlier work, we explored glycine- and serine-rich octapeptides using molecular dynamics (MD) simulations in combination with concepts from FRET (Förster Resonance Energy Transfer) spectroscopy. The goal was to understand how flexible chains fluctuate and how these fluctuations are reflected in experimentally measurable distances.
In a new publication in The Journal of Physical Chemistry B [1], we have built directly on that foundation, but push the idea further toward quantitative integration between simulation and experiment. At the center of both studies is a small but powerful fluorescent probe: 2,3-diazabicyclo[2.2.2]oct-2-ene (DBO). Paired with tryptophan, DBO enables measurements of extremely short intramolecular distances. Because it is compact and minimally perturbing, it is particularly well suited for probing flexible peptides that would be difficult to characterize using larger fluorophores. In the earlier work, the focus was primarily on understanding conformational ensembles and distance distributions.
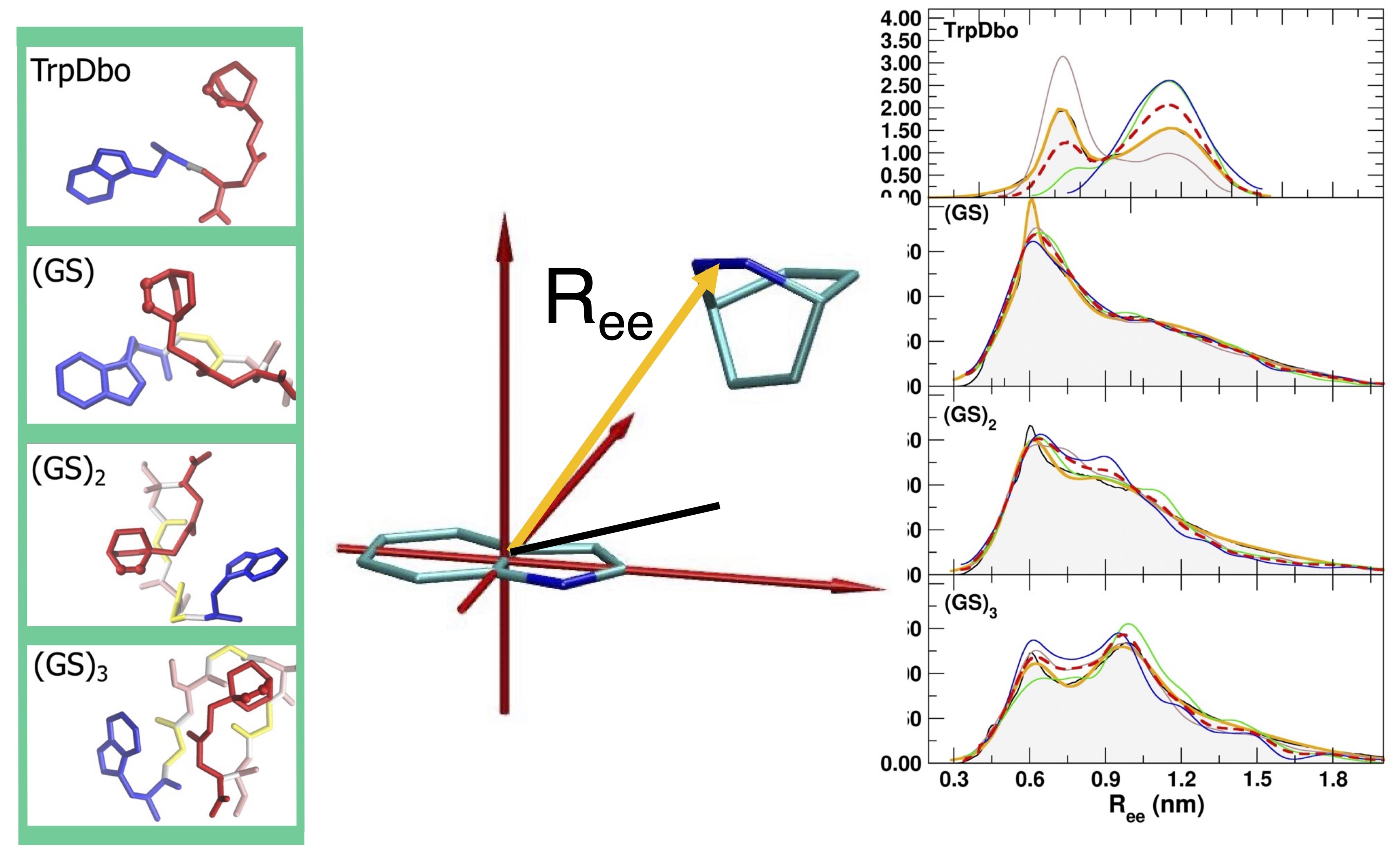
In this new study, the Dbo model has been upgraded to the more recent version of the GROMOS force field (54A7), and using extensive MD simulations, we have verified whether the new model can more quantitatively reproduce both structural and kinetic FRET experimental observables. In particular, we combined time-resolved FRET experiments with microsecond-scale MD simulations to study model peptides of the form Trp–(GS)n–Dbo and Trp–(PP)n–Dbo with n=0,1,2,3, where the glycine–serine sequences represent highly flexible chains, and the polyproline sequences provide a more rigid reference.
The results of the simulations showed:
- Simulated end-to-end distances agree with FRET-derived experimental values within 5% for the flexible (GS)_npeptides.
- Contact formation kinetics (looping rates) quantitatively match experiment once solvent viscosity is properly accounted for.
- The relationship between chain flexibility and fluorophore separation is systematically captured.
Beyond equilibrium averages, we also analyzed time correlations and dynamical fluctuations, linking conformational free-energy landscapes to experimentally observable FRET signals.
Instead, it demonstrates that combining equilibrium FRET distances and time-resolved kinetic data provides a stringent benchmark for simulation models of flexible peptides. Furthermore, this integrated FRET–MD framework with the improved Dbo model can be applied to:
- Flexible linkers in multidomain proteins
- Intrinsically disordered protein segments
- Small proteins undergoing conformational adaptation
REFERENCE
[1] D. Roccatano . Quantitative Integration of FRET and Molecular Dynamics for Modeling Flexible Peptides. J. Phys. Chem. B, (2026-02-27)
doi: https://doi.org/10.1021/acs.jpcb.5c08148








 , where N is the total number of electrons in the system. It was clear that a reduction, using ad hoc approximations, of the description of the dynamic behaviour of atoms using a classic physics model would be necessary to overcome this problem. In the classical representation, the electrons on the atoms are not explicitly considered, but their mean-field effect is taken into account. Alder and Wainwright performed the first simulation of an atomic fluid using this approximation approximately 63 years ago (1957). They developed and used the method to study simple fluids by means of a model representing atoms as discs and rigid spheres. These first pioneer studies mark the birth of the classical molecular dynamics (MD) simulation technique. The successive use of more realistic interaction potentials has allowed obtaining simulations comparable to experimental data, showing that MD can be a valuable tool for surveying the microscopical properties of physical systems. The first simulations of this type were carried out by Rahman and Verlet (1964): in these simulations, a Lennard-Jones-type potential was used to describe the atomic interactions of argon in the liquid state. Another significant hallmark in this field was the simulation of the first protein (the bovine pancreatic trypsin inhibitor) by McCammon and Karplus in 1977. In the following years, the success obtained in reproducing structural properties of proteins and other macromolecules led to a great spread of the MD within structural biology studies. The continuous increase of computer power and improvement of programming languages has concurred with further refinement of the technique. Its application was progressively expanded to more complex biological systems comprising large protein complexes in a membrane environment. In this way, MD is becoming a powerful and flexible tool with applications in disparate fields, from structural biology to material science.
, where N is the total number of electrons in the system. It was clear that a reduction, using ad hoc approximations, of the description of the dynamic behaviour of atoms using a classic physics model would be necessary to overcome this problem. In the classical representation, the electrons on the atoms are not explicitly considered, but their mean-field effect is taken into account. Alder and Wainwright performed the first simulation of an atomic fluid using this approximation approximately 63 years ago (1957). They developed and used the method to study simple fluids by means of a model representing atoms as discs and rigid spheres. These first pioneer studies mark the birth of the classical molecular dynamics (MD) simulation technique. The successive use of more realistic interaction potentials has allowed obtaining simulations comparable to experimental data, showing that MD can be a valuable tool for surveying the microscopical properties of physical systems. The first simulations of this type were carried out by Rahman and Verlet (1964): in these simulations, a Lennard-Jones-type potential was used to describe the atomic interactions of argon in the liquid state. Another significant hallmark in this field was the simulation of the first protein (the bovine pancreatic trypsin inhibitor) by McCammon and Karplus in 1977. In the following years, the success obtained in reproducing structural properties of proteins and other macromolecules led to a great spread of the MD within structural biology studies. The continuous increase of computer power and improvement of programming languages has concurred with further refinement of the technique. Its application was progressively expanded to more complex biological systems comprising large protein complexes in a membrane environment. In this way, MD is becoming a powerful and flexible tool with applications in disparate fields, from structural biology to material science.




