“If you know the enemy and know yourself, you need not fear the result of a hundred battles. If you know yourself but not the enemy, for every victory gained you will also suffer a defeat. If you know neither the enemy nor yourself, you will succumb in every battle.”
SunTzu. The Art of War
A virus is the Bauhaus of the form of life: the minimalist reduction of an organism to its essential element of functionality. More pragmatically, it is a container of genetic code provided with a smart mechanism that allows it to invade cells of another host organism. As a molecular machine, a virus can resemble in shape and destructive power the Death Star spaceship of the Star War saga. Therefore, it is a molecular machine that we do not definitively want to have within us!
The spread of the coronavirus SARS-CoV-2 has produced a new pandemic, i.e. an infection caused by a pathogen that affects the entire population of a living species, in this case the human one. This global emergency situation is the result of a natural competition between living species that reminds us that we are still a small brick of the Gaia ecosystem. However, although it is always difficult to believe given the state in which we have reduced our planet, we are the most intelligent life form in the known universe. So it would be quite embarrassing to be defeated by an invisible enemy.
As the old Chinese sapience of Sun Tzu suggest: the best way to find the enemy is to know it (and ourselves). This war has urged me to start this blog where I will share what I am learning about this dangerous microscopic f o e (=form of existence) from the perspective of my scientific interests.
The coronaviruses are a group of zoonotic viruses having as host mammals and birds. They have strategically developed a high mutability rate to survive the immune defences of host organisms. They enter the host through the tissues of the airways and are therefore easily transmitted. The high mutability of the genetic material of this type of virus, the high similarity of the immune system of the infested species (such as pigs, mice, bats, camels or birds), and their close contact with humans, have determined (and, on some occasions in a time shorter time than the average duration of human life) transmission via interspecies jumps. In this case the new virus has become a dangerous new threat to the human species. In late November 2019, one of this jump between an unidentified sick animal and its seller and/or buyer, occurred in an animal and seafood market in the city of Wuhan (China). Officially, this marked the beginning of the spread of a new potentially lethal human coronavirus species. It is still unclear the species of animal that transmitted the virus to humans. Bats are among the candidates, but more recent studies also suggest pangolins.
A virus that becomes host to another species can be very dangerous for the latter. In fact, in the species that originally carriers the virus, the natural selection has adapted the immune system to cope with the invasion of the virus. Similarly to what our immune system manages to counteract the common flu. On the contrary, the immune system of the new host species may either not notice the invader responding too slowly, or producing an excessive response to the infection. In the latter case, the immune defence mechanism itself can become the cause of the danger. This situation appears to be one of the main causes that makes COVID-19 fatal. Many of us, especially the most vulnerable because they are suffering from age-related complications, or with a compromised respiratory system, are overwhelmed by the counter-offensive of the immune defences which instead of saving them ends up drowning them. Much of the death toll from COVID-19 is the sad consequence of this effect.
To try to understand what is happening at the molecular level, I have to summarize what is known about the structure of this complex nanoscopic war machine and its invasion and reproduction mechanism in its guests.
The life cicle of a virus comprises more or less the following steps:
- The virus attaches itself to its host cell. In the case of the CORVID-19 this occurs in to the receptors on the epithelium cell of the respiratory tract of the lungs.
- The virus penetrates the cell, for the COVID-19, the lipid envelope fuse with the cell lipid membrane.
- The nucleic acid (RNA for the COVID-19) is uncoated (the capsomere made of nucleocapsid proteins dissociate) and ready to be read by expression machinery of the host cell.
- Normally at this stage only part of the genetic code is expressed to this stage contributing to part of the genetic function such as the replication of the viral chromosome. In same case, it can also turn off function of the host cell to maximize the cell’s available resources for the virus production.
- The virus produce hundreds copies of its genetic material.
- At this stage, the virus start to produce the structural protein for capsomere and other proteins embedded in the external lipid membrane.
- The nucleocapsid proteins assemble around the RNA of the virus forming the capsomere. The three membrane proteins of the virus are transported on the external membrane of the host cell.
- The capsomere is finally released by the cell by exocytosis by coating it with a piece of cell membrane populated with virus proteins.
I have added at the end of this post link to interesting animations that visually summarize all the process.
To understand the detail of each stage of this complex process, we need to dissect and analyse in detail (or at the least what it is known about) of the structure and function of a coronavirus.
In general, viruses are particles with size ranging from 0.02 to 0.25 micrometre (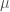
Depending on the type of genetic material they carry, they are classified as DNA and RNA type virus. The difference also determines the kind of mechanism they exploit to reproduce themselves once inside the host cell. RNA and DNA virus can integrate their DNA in the DNA of the host species. It is even hypothesized that this mechanism was responsible for the evolution of primordial prokaryotic to the more complex eukaryotic cell. RNA’s viruses such as the nCovid-19 do not translate their RNA in DNA, but they use directly the cell equipment (ribosome) to reproduce themselves. This makes them more virulent but maybe less subtle in their camouflage.
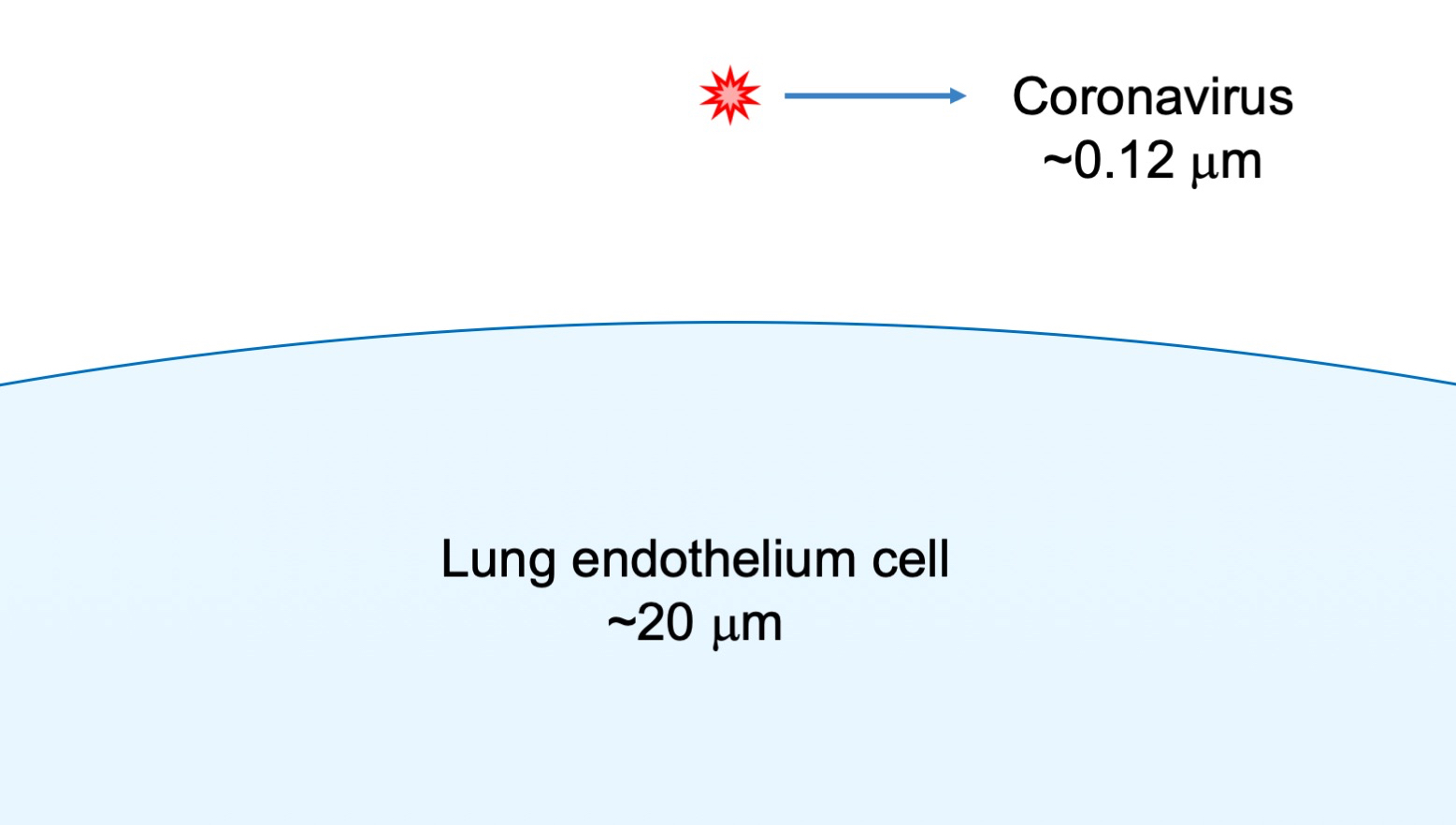
The genome of coronaviruses has a size ranging between ~26000 and ~32000 nucleic bases, and codify for a variable number (from 6 to 11) of protein sequence. The coronavirus nCovid-19 contains 30 kilobases, the first and also the longest part of this sequence (representing approximately 67% of the entire genome) encodes 16 non-structural proteins, while the rest accessory proteins and structural proteins. In Figure 2, it is reported a schematic description of the COVID-19 genome. The structure of several proteins is still not determined and the function not well clarified.
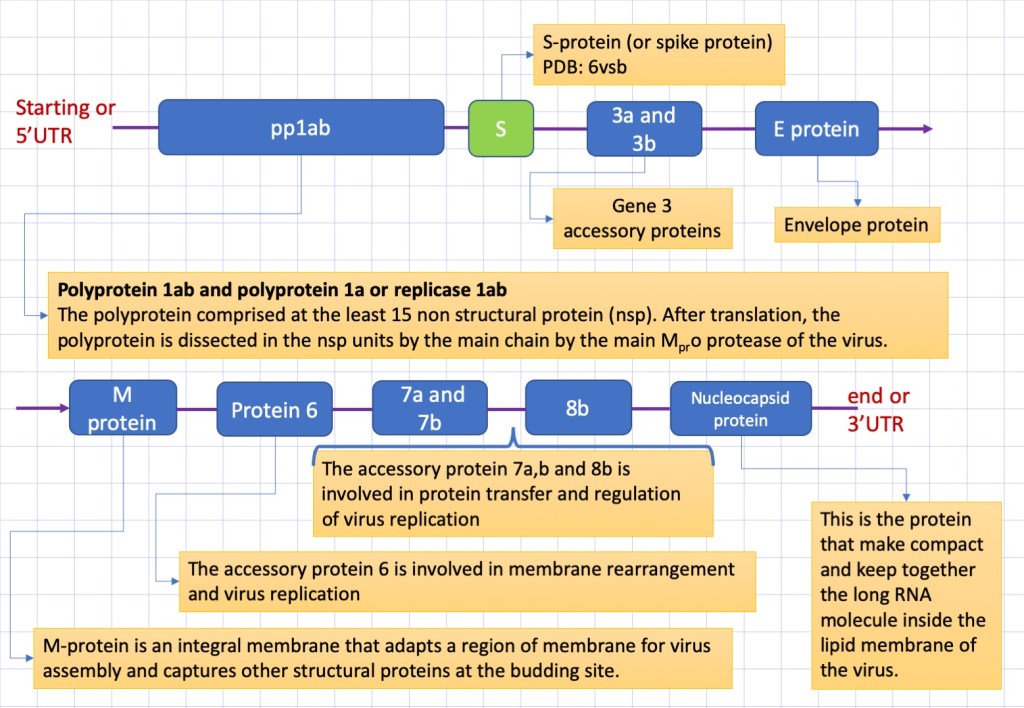
We start to analyse the structural part of the enzyme starting from the external envelop. This is made by lipids, the same that compose the eukariotic cell membrane. Inserted to the membrane there are the three major structural proteins: the S-protein a glycoprotein protruding from the membrane and responsable to the binding to the cell receptor of the host cell, the small envelope protein (E) and matrix protein (M). Let see in detail the nature and function of these proteins.
The S-protein (S is for surface) or spike protein is the most essential virus membrane glycoprotein. It plays the crucial role in binding to receptors on the host cell induce the fusion of the virus membrane with the one of the host cell. The process trigger the invasion of the host cell. Given its importance, the S-protein is one of the primary target for drugs against the virus. The structure of the pre-fusion trimer of the S-protein has been recently solved using cryo-electron microscopy. From a collected set of images, it has been possible to reconstruct the three dimensional structure of the protein at atomic level. The molecular models of the protein available in the protein data base (PDB codes: 6vsb, 6vxx, 6vyb).
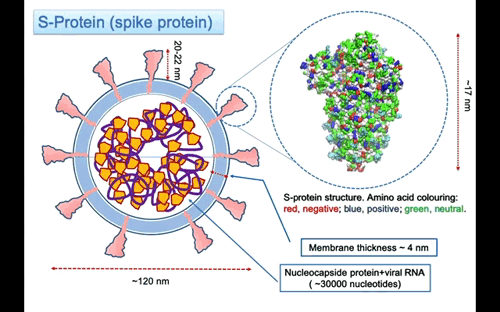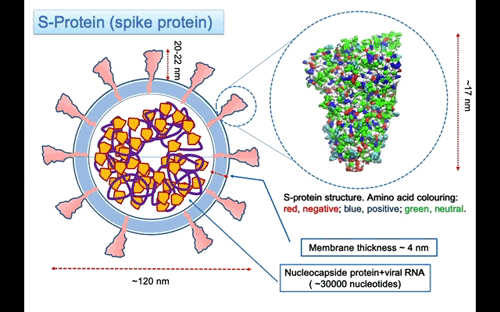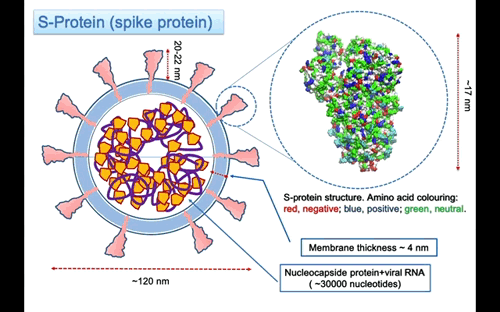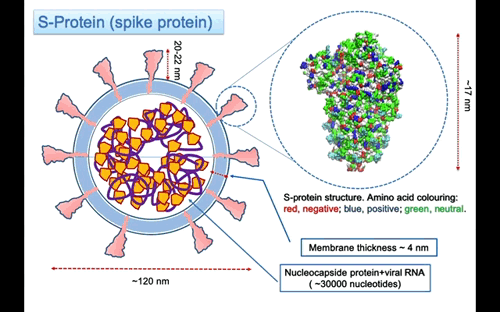
In Figure 3, the structure of the S-protein is represented using van der Waals sphere for the atoms coloured according to the polar nature of the amino acid, as red negative, blue positive and green neutral type. The protein is ~17 nm long, but the three helices stalk connecting with the membrane is missing. I might expect that the total lenght of the protein to the membrane surface is around 20-22 nm. The three helices insert into the membrane and protruding in the interior part with a region probably anchored to the nucleocapsid proteins that surround the RNA molecule. On the largest top part the spike is 12×12 nm large. The protein is a glycoprotein, this mean that some of the residue on the surface are attached to glycan that are polysaccharides composed of different types of type of monosaccharides units forming linear or branched chains.
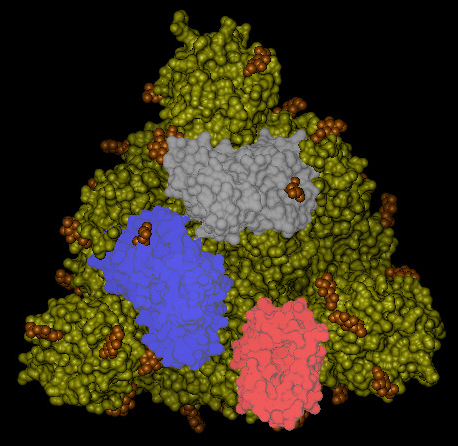
Glycans perform a fundamental function in the mechanism of molecular recognition of receptors on the surface of cells in our body. This function is exploited by viruses to camouflage and evade our immune system. Fortunately, the glycans that cover the proteins on the outer membrane of these viruses are generally less complex than those present in the host organism in this way our immune system learns, even if more slowly, to “distinguish” the sugar canopy forest on the viruses and to neutralize them. A recent article published in the American journal Science [5] reports a detailed mapping of glycans on the surface of the Spike protein. This is a very important result as it will allow more effective vaccine development. In the resolved structure of protein S, sixty six N-glycosylation sites (22 per chain) are reported. In the structure the amino acids are linked to single N-acetyl-glucosamine molecules (NAG, see structural formula in Figure 4) but the article in Science has shown a detailed map of the sites that are glycosylated with sugar chains of different complexity and composition.
The NAG units looks quite uniformly distributed all around the S-protein trimer (see Figure 5).
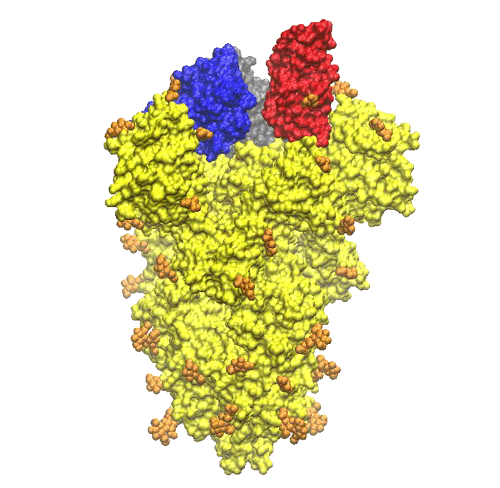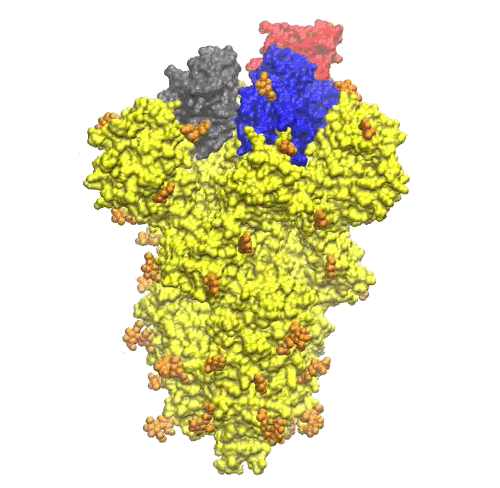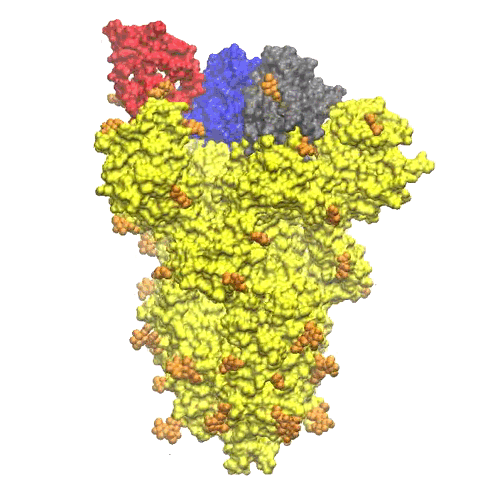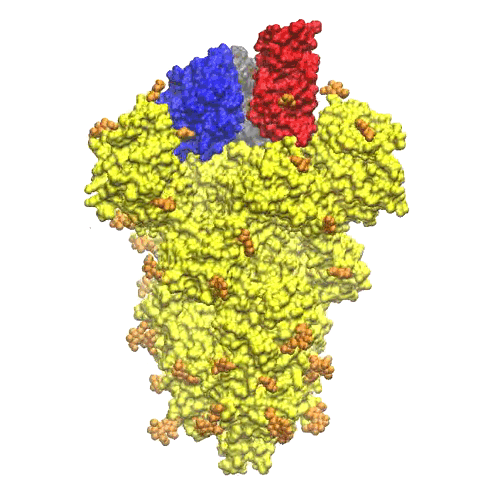
On the top of the spike, there are three regions that are identified as receptor binding domains (see Figure 6).
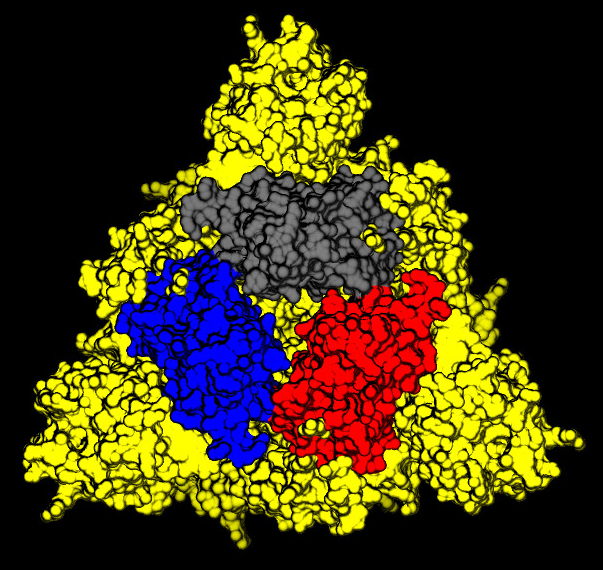
The domains undergoes an open/closing motion around a hinge evidenced by the difference observed between the PDB structures 6vxx (closed, Figure6) and 6vvy (open, Figure7), respectively. In Figure7, the open domain is in red and it orientation is visible also in Figure 5.
The RDB are involved in the binding to the receptor that in the case of the COVID-19 has been identified as the Angiotensin-converting enzyme 2 (or ACE2). This is an enzyme attached to the outer surface (cell membranes) of cells in the lungs (but also arteries, heart, kidney, and intestines). ACE2 lowers blood pressure by catalysing the hydrolysis of angiotensin II (a vasoconstrictor peptide) into angiotensin (a vasodilator).
ACKNOWLEDMENTS
All the figures of the protein structures in this article have been produced using the public domain software Visual Molecular Dynamics (VMD) developed at the University of Illinois at Urbana – Champaign [6].
REFERENCES AND OTHER INFORMATION MATERIAL
- Wrapp, D., Wang, N., Corbett, K.S., Goldsmith, J.A., Hsieh, C.L., Abiona, O., Graham, B.S. and McLellan, J.S., 2020. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science, 367(6483), pp.1260-1263.
- Levine, A.J., 1991. Viruses: A Scientific American Library Book. Henry Holt and Company.
- Wu, A., Peng, Y., Huang, B., Ding, X., Wang, X., Niu, P., Meng, J., Zhu, Z., Zhang, Z., Wang, J. and Sheng, J., 2020. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell host & microbe.
- Yan, R., Zhang, Y., Li, Y., Xia, L., Guo, Y. and Zhou, Q., 2020. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science.
- Watanabe, Y., Allen, J.D., Wrapp, D., McLellan, J.S. and Crispin, M., 2020. Site-specific glycan analysis of the SARS-CoV-2 spike. Science.
- Humphrey, William; Dalke, Andrew; Schulten, Klaus (February 1996). “VMD: Visual molecular dynamics”. Journal of Molecular Graphics. 14 (1): 33–38
Userful and Interesting Videos
Nucleus Medical Media: https://www.youtube.com/watch?v=5DGwOJXSxqg
RCSBProteinDataBank: https://www.youtube.com/watch?v=s2EVlqql_f8
Scientific Animations: https://www.youtube.com/watch?v=I-Yd-_XIWJg&t=143s
Biolution: https://www.youtube.com/watch?v=I0TmBsHaGmI

