Atomic Fractional Coordinates
Atomic fractional coordinates (AFCs) are used to specify the positions of atoms within a crystal structure. They are expressed as three fractional values 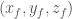
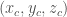
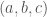
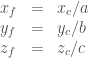
therefore, the value of ACFs are fractional values between 0 and 1. For example, an atom at (0.25, 0.5, 0.75) is located a quarter of the way along the a-axis, halfway along the b-axis, and three-quarters of the way along the c-axis of the unit cell. Crystal structures often exhibit symmetry, and fractional coordinates are essential for understanding and describing this symmetry. Symmetry operations, such as rotations, translations, and reflections, can be applied to fractional coordinates to elucidate the symmetrical aspects of the crystal, which is crucial for understanding the material’s properties.
Conversion of ACFs to Cartesian Coordinates in a Triclinic Lattice
Converting atomic fractional coordinates to Cartesian coordinates is done using the lattice parameters (a, b, c) and the angles between them (α, β, γ). For ortogonal lattices, the formula

However, if you’re dealing with non-rectangular unit cells (α ≠ 90°, β ≠ 90°, γ ≠ 90°), you’ll need to apply a more general formula that account for the angles between lattice vectors. The formula, expressed in matrix form is the following

These additional trigonometric terms adjust for the angles between lattice vectors, making the conversion appropriate for triclinic systems. For (α = 90°, β = 90°, γ =90°), this expression reduce to the simple one for the ortogonal lattices.
Atom coordinates of structure of small molecules resolved using X-ray crystallography are normally provides as fractional coordinates are often included within crystallographic data files, such as CIF (Crystallographic Information File) and certain specialized crystallography data file formats. These files contain information about the crystal structure, including unit cell parameters, atomic positions, and other crystallographic data, and they may represent atomic positions using fractional coordinates.
CIF (Crystallographic Information File): CIF is a commonly used file format in crystallography for storing crystallographic information, including atomic positions in fractional coordinates. CIF files provide a structured way to represent crystallographic data, making them well-suited for sharing and exchanging information about crystal structures.
Here’s an example of CIF file containig the molecule of UREA[1]. The file was obtained from the Open Crystallographic Database.
data_1008776 _symmetry_cell_setting tetragonal _symmetry_space_group_name_H-M 'P -4 21 m' _cell_length_a 5.661 _cell_length_b 5.661 _cell_length_c 4.712 _cell_angle_alpha 90 _cell_angle_beta 90 _cell_angle_gamma 90 _symmetry_equiv_pos_as_xyz x,y,z 1/2-x,1/2+y,-z -x,-y,z 1/2+x,1/2-y,-z -y,x,-z 1/2+y,1/2+x,z y,-x,-z 1/2-y,1/2-x,z loop_ _atom_site_label _atom_site_type_symbol _atom_site_symmetry_multiplicity _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z C1 C4+ 2 0. 0.5 0.333(1) O1 O2- 2 0. 0.5 0.5968(11) N1 N3- 4 0.1439(9) 0.6439(9) 0.1832(5) H1 H1+ 4 0.2522(26) 0.7522(26) 0.2839(21) H2 H1+ 4 0.1365(19) 0.6365(19) 0.9724(14)
In this example, the Data Block Header (data_1008776) indicates the beginning of a new data block with the identifier “1008776.” Then, the CIF file contains symmetry Information about the unit cell parameters (_cell_length_a, _cell_length_b, etc.) and smmetry equivalents that provides equivalent positions in fractional coordinates to define the crystal symmetry. The fractional coordinates of oxygen (O), nitrogen (N) and hydrogen (H) atoms are represented in the _atom_site_fract_x, _atom_site_fract_y, and _atom_site_fract_z fields.
Fractional coordinates are essential in crystallography because they account for the unit cell’s periodic nature and are independent of the specific unit cell dimensions, making it easier to compare structures with different unit cell sizes.
XYZ Format (Cartesian Coordinates):
The XYZ format is a simple and widely used format for storing atomic coordinates in Cartesian coordinates (x, y, z). Each line in the file typically represents an atom and contains the following information:
- Atom symbol: The chemical symbol of the atom.
- x-coordinate: The x-coordinate of the atom in Ångströms.
- y-coordinate: The y-coordinate of the atom in Ångströms.
- z-coordinate: The z-coordinate of the atom in Ångströms.
For example, here’s how an XYZ file might look:
3
Mg 0.000 0.000 0.000
O 1.000 0.000 0.000
O -1.000 0.000 0.000
The first line indicates the number of atoms in the system, followed by lines for each atom, specifying its element and coordinates.
PDB Format (Protein Data Bank Format):
The PDB format is commonly used to store atomic coordinates for biomolecules (proteins, DNA, RNA) and small organic molecules. PDB files are more structured and include additional information beyond atomic coordinates, such as atom connectivity, residue information, and more. Here’s an overview of the format:
- ATOM records: These records specify atomic coordinates and are structured as follows:
- Atom serial number.
- Atom name.
- Residue name.
- Chain identifier.
- Residue sequence number.
- Coordinates (x, y, z) in Ångströms.
- Atomic occupancy and temperature factor.
- Element symbol.
In the following example the atomic coordinates of the Glycine molecule is reported
HEADER AMINO ACID 01-MAR-2023 ATOM 1 N GLY A 1 1.000 2.000 3.000 1.00 15.00 N ATOM 2 CA GLY A 1 1.000 3.000 3.000 1.00 15.00 C ATOM 3 C GLY A 1 2.000 4.000 3.000 1.00 15.00 C ATOM 4 O GLY A 1 2.000 5.000 3.000 1.00 15.00 O ATOM 5 OXT GLY A 1 3.000 4.000 3.000 1.00 15.00 O
Explanation:
- HEADER line: Provides general information about the entry.
- ATOM lines: Describe the atomic coordinates. In this example:
- The OXT atom is often used in PDB files to represent the oxygen atom of the terminal carboxyl group, providing a complete representation of the amino acid structure within a peptide or protein chain.
In the following example the atomic coordinates of the Urea molecule is reported
HEADER Title: Urea REMARK CRYST1 29.70 34.30 15.10 90.00 90.00 90.00 P 1 HETATM 1 C1 URE 1 20.640 21.620 2.800 1.00 0.00 HETATM 2 O2 URE 1 21.630 21.200 3.390 1.00 0.00 HETATM 3 N3 URE 1 20.550 22.850 2.290 1.00 0.00 HETATM 4 H4 URE 1 19.650 23.010 1.880 1.00 0.00 HETATM 5 H5 URE 1 21.360 23.420 2.120 1.00 0.00 HETATM 6 N6 URE 1 19.510 20.930 2.850 1.00 0.00 HETATM 7 H7 URE 1 18.660 21.250 2.440 1.00 0.00 HETATM 8 H8 URE 1 19.580 19.990 3.200 1.00 0.00 CONECT 1 2 CONECT 1 3 CONECT 3 4 CONECT 3 5 CONECT 6 1 CONECT 6 7 CONECT 6 8 TER END
- HETATM records in PDB files is a way to distinguish non-standard residues or small molecules from standard amino acids.
- The TER line indicates the end of the urea molecule.
- The END line marks the end of the PDB file.
- CONECT records specify connectivity between atoms. Each line starts with the atom serial number, followed by a list of atom serial numbers that the given atom is connected to.
While PDB files are commonly associated with biomolecules, they can be used for any crystallographic structure. Many visualization and molecular modeling software tools can read and write PDB files. These formats serve as standards for storing atomic coordinates, making it easy to share and exchange structural data among researchers and visualization software. Depending on your needs and the software you are using, you may choose one format over the other.
WHERE TO FIND THE CRYSTAL STRUCTURE COORDINATES
If you’re looking for crystallographic data of molecules, you can explore the following databases:
- Cambridge Structural Database (CSD): Contains structural data for organic and metal-organic compounds. It’s one of the most extensive databases for small molecule crystal structures.
- Inorganic Crystal Structure Database (ICSD): Focuses on inorganic and metal-organic crystal structures.
- Protein Data Bank (PDB): While primarily for biological macromolecules, it includes many small organic compounds as ligands in complex structures.
- American Mineralogist Crystal Structure Database (AMCSD): Concentrates on mineral crystal structures.
- Crystallography Open Database (COD): A free and open-access resource that includes crystal structures of organic, inorganic, and metal-organic compounds.
These databases provide access to a wealth of crystallographic data that can be used for research and analysis. Research articles published in scientific journals often include crystallographic data and details of crystal structures. Journals like Acta Crystallographica, Journal of Applied Crystallography, and Inorganic Chemistry are good sources for finding crystal structure information.
REFERENCES
- Worsham, J.E., Levy, H.A. and Peterson, S.W., 1957. The positions of hydrogen atoms in urea by neutron diffraction. Acta Crystallographica, 10(4), pp.319-323.

